
A Primer on Molecular Docking
Why docking scores don’t predict binding affinity, and what template-guided approaches actually deliver

Welcome back to Kiin Bio Weekly.
For computational chemists, drug discovery scientists, and anyone running or interpreting docking campaigns.
Docking scores do not predict binding affinity. The OpenBind benchmark confirmed it: molecular weight alone outperformed Gnina, Boltz-2, and other state-of-the-art models on experimental binding data.
The real value of docking is pose prediction, not ranking compounds. Most teams using it well have stopped asking it to estimate affinity altogether.
This piece covers where docking fails, where it works (template-guided lead optimisation), and why pooling imperfect methods beats waiting for a perfect one.
Freebie alert: We know how hard science is. That’s why we built the Pioneer Programme.
We’re selecting academic and nonprofit teams to get one year of free access to our drug discovery platform, with support from our science team. If you spend more time pulling together findings from different sources than actually acting on them, it’s worth applying.
No cost, no data transfer, all IP stays with your institution. Applications close August, cohort starts September.

For this piece, I spoke with Rachael Skyner, our cheminformatician at Kiin Bio, about the mechanics of molecular docking, why scoring remains unsolved, and how template-guided approaches are changing what is possible in lead optimisation. Her insight runs throughout.
“The search problem is not the hard part. It’s ranking them and picking the right pose.”
Why this matters now
Thanks to AlphaFold and its successors, drug discovery teams now have predicted structures for essentially every human protein. Generative chemistry tools are producing novel molecules faster than medicinal chemists can evaluate them. Having a structure to dock into is no longer the problem. Trusting what the docking tells you is. The OpenBind benchmark, published earlier this year, put hard numbers on this trust deficit for the first time, and the results were not encouraging.
Docking can tell you roughly where a molecule might land. It cannot reliably tell you how tightly it will stick. Most practitioners have adapted by splitting the problem in two: get the pose right first, worry about affinity later. That distinction shapes everything about how docking is actually used today.
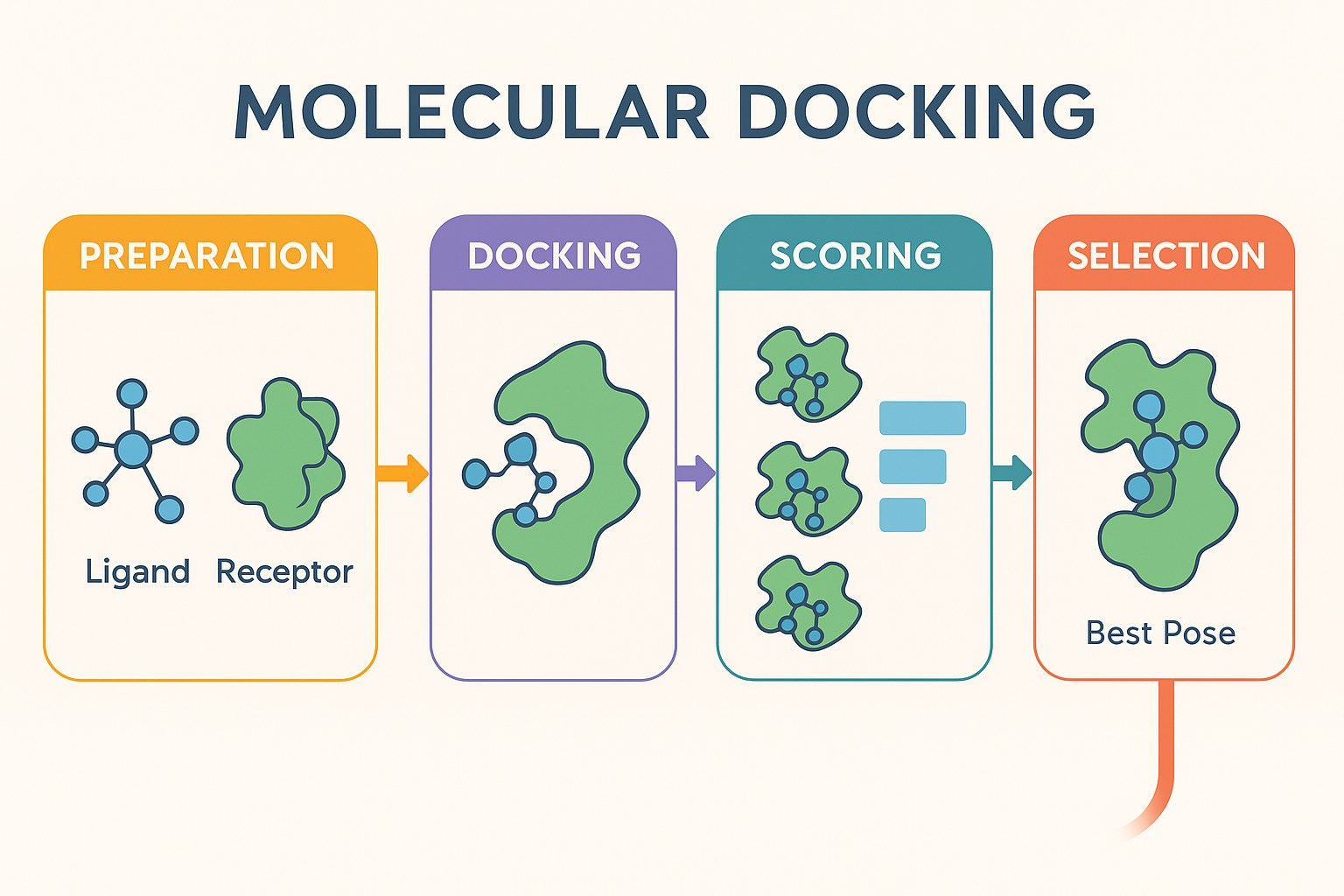
The scoring problem nobody has solved
Traditional docking scores are supposed to estimate binding affinity. They do not. The physics approximations used in scoring are rough even for a static system, and on top of that, docking ignores the fact that binding is actually a dynamic process. Binding in the body involves solvent reorganisation, protein conformational changes, entropy costs, and timescale-dependent interactions that no static pose can capture.
“It’s an oversimplification of what’s actually going on with binding,” says Skyner. “It’s not just binding that contributes to affinity in the end. There are lots of other things that can influence how strongly a molecule binds. You’re treating it as a static problem when in reality it’s very dynamic.”
Increasingly, people are separating pose prediction from affinity estimation altogether. Modern scoring methods from tools like Gnina use convolutional neural networks trained on the PDBbind dataset. Rather than predicting binding affinity directly, the CNN score predicts confidence in whether the pose is correct, which is a more tractable and more useful question to ask.
Protein flexibility and the induced fit problem
Traditional rigid docking assumes the protein does not move, which is obviously wrong. Proteins reshape themselves around ligands in a process called induced fit, and this is why rigid receptor docking often fails for novel chemotypes.
Physics-based approaches like Rosetta and Schrodinger’s IFDMD allow limited residue rotation or flipping at the binding site. Machine learning methods like DiffDock take a generative approach, producing the molecule directly into the protein. Co-folding methods fold the protein and ligand simultaneously, addressing flexibility implicitly by allowing both to move at the same time during prediction.
The problem is that induced fit methods produce multiple protein conformations alongside multiple ligand conformations, and the analysis becomes exponentially more complex. “From an analysis point of view, it’s really difficult to deal with those structures,” says Skyner. “You have to start doing more complicated things like clustering together both the ligand and the protein conformation to see which ones come up most often.”
For high-throughput virtual screening where you want to process hundreds of thousands of molecules, this complexity is impractical. Induced fit docking belongs in the later stages of a project, when you already have a binder and want hypotheses about its binding mode.
Traditional rigid docking assumes the protein does not move. Proteins move constantly. They reshape themselves around ligands in a process called induced fit, and this is why rigid receptor docking often fails.
The virtual screening funnel
In practice, docking is rarely used alone. It sits inside a virtual screening funnel: start with a compound library (thousands to millions of molecules), run fast docking as a heuristic filter (anything scoring worse than minus eight kilocalories per mole gets discarded), then rescore the surviving hits with more expensive methods.
Rescoring typically involves energy minimisation with solvent. Methods like MMGBSA and MMPBSA incorporate water into the calculation, which basic docking ignores entirely. “When you start thinking about the solvent as well, you start getting much better correlation with experimental affinities,” says Skyner. These methods are computationally expensive, though still feasible for the few hundred molecules that survive initial filtering.
ML-based rescoring adds another option: build a local model of the docking score and then only dock the subset your model flags as likely to score well. This can cut the number of molecules you need to physically dock in half.
Where docking actually works: template-guided lead optimisation
Docking works best in template-guided approaches during lead optimisation, not in blind global searches or massive virtual screens. This is where Skyner’s work focuses.
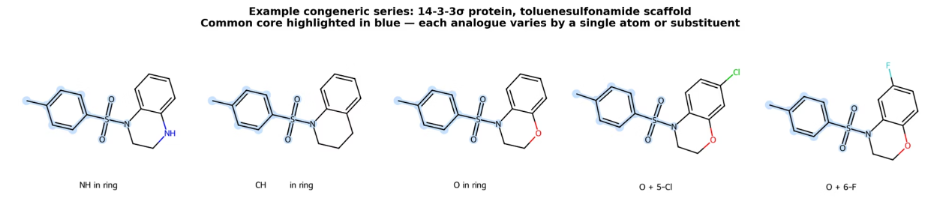
At this stage, you already have a molecule with known binding evidence. You are working through a congeneric series: molecules that share a common core (the maximum common substructure, or MCS) with small modifications around the periphery. A chlorine added here, a fluorine swapped there, an atom changed in a ring. You already know the molecule binds. What you want to understand is whether the modification changes how it sits in the pocket.
Template-guided docking uses the known crystal structure of a reference compound to anchor the search. Rather than exploring the entire binding pocket from scratch, you start from the position of the known binder and run a local minimisation. Skyner has developed an MCS-guided docking approach that generates low-energy conformers of the ligand, rigidly aligns them to the reference structure via the MCS, and performs local optimisation from that aligned position using AutoDock Vina.
“If you give it the right starting position, it’s got a much better chance of getting the pose correct,” says Skyner. “People might do this by default, but it really, really helps in these scenarios.”
Ensembles, pooling, and what comes next
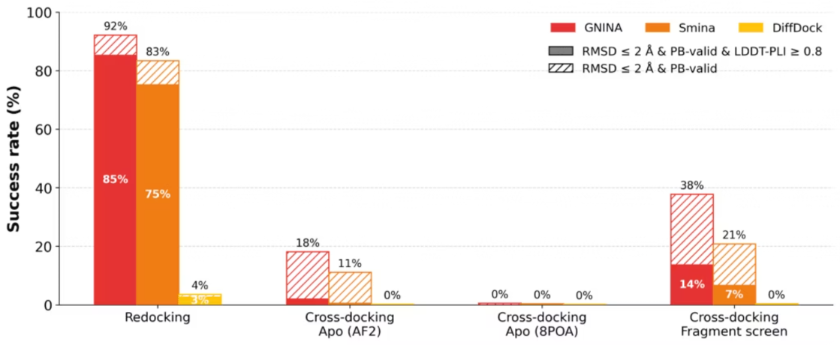
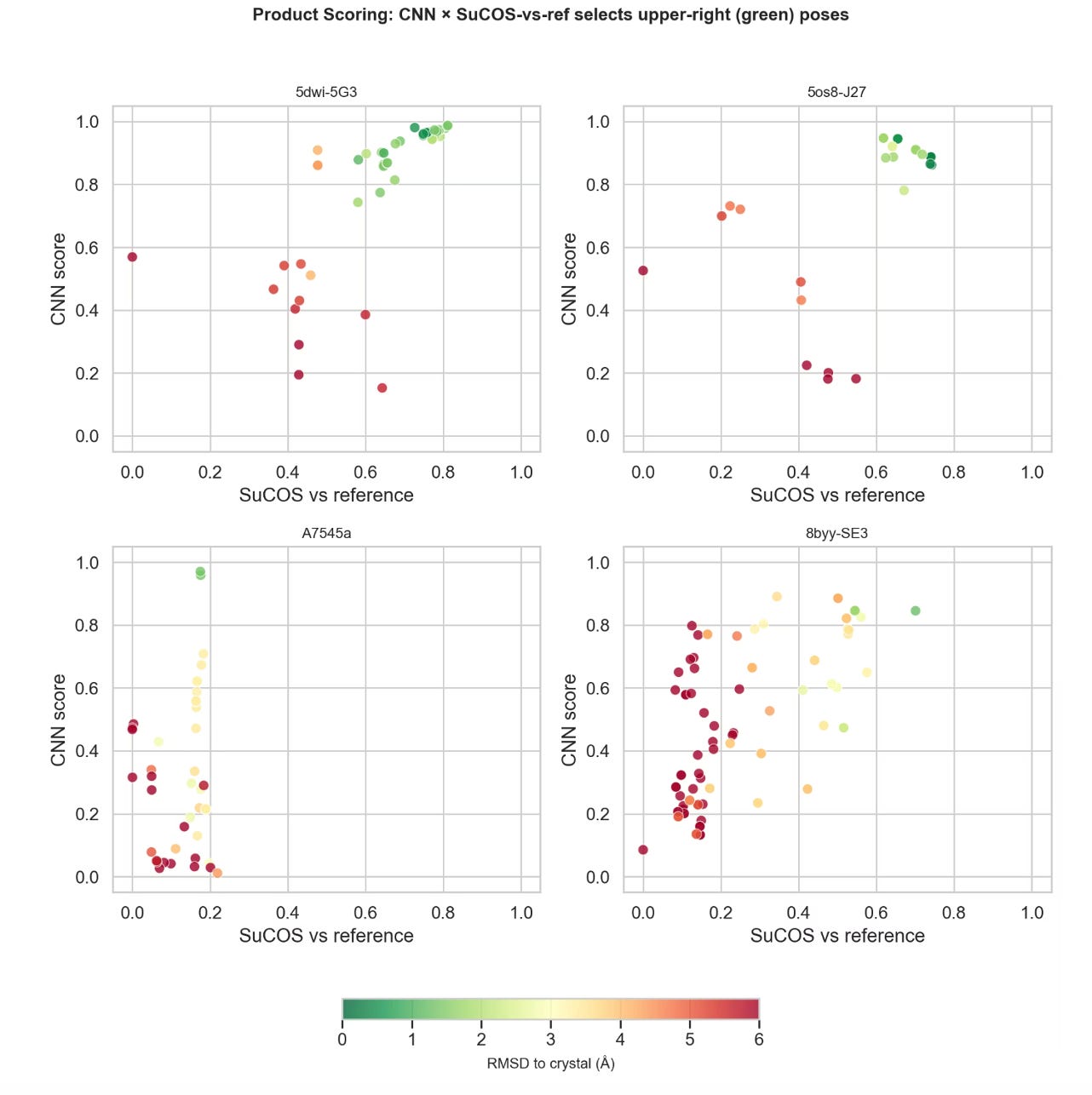
No single docking method dominates across all targets. In Skyner’s evaluation of 22 targets (1,888 structures), pooling results from multiple methods and rescoring with Gnina’s CNN score consistently outperformed any individual approach. The pooled “oracle” (checking whether any method generated the correct pose) found it 90% of the time using RMSD <2 angstroms as the heuristic for a correct binding pose, and 54% by the more stringent SuCOS metric, which evaluates shape and pharmacophore overlap rather than just atomic positions.
The practical recommendation: combine Vina (fast, global search) with MCS-guided Vina (template-aware), pool the poses, and rescore with the CNN score. Two methods captured nearly all the benefit of three, with substantially less compute.
The preparation of proteins and ligands is still the most error-prone step in any docking workflow. Get the protonation states wrong, miss a stereoisomer, assign incorrect charges, and the docking algorithm has no chance of finding the right answer. Errors compound downstream into molecular dynamics and other physics-based follow-up methods. “If you get it wrong from the beginning, it really affects everything else,” says Skyner.
Docking will not predict your binding affinities, and it will not reliably rank compounds within a series. The industry spent years asking it to do both, and the benchmarks now confirm what practitioners quietly knew. The gains are coming from asking docking the right question in the right context: use it for pose prediction rather than affinity estimation, use template-guided exploration rather than blind search, pool results from multiple methods rather than betting on one. The teams actually getting value out of docking today have mostly just stopped asking it to be something it was never designed to be.
💬 Want to be featured in Kiin Bio Weekly?
Each issue we speak directly with researchers, scientists, and builders working at the frontier of AI in life sciences. If you're working on something in this space and think it would resonate with our community, I'd love to hear from you. Fill out this form or reach out to me directly.
Found this useful? Forward it to a colleague in computational biology, it's the best way to help the newsletter grow.
Subscribe now to stay at the forefront of AI in Life Science. Every week: primers, deep dives, and direct conversations with the people building the field.
Connect With Us
Have questions on this or suggestions for our next deep dive? We’d love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website