
🧪A Primer on Peptide Cheminformatics
The representations, fingerprints, and AI models being built for peptide drug discovery

Welcome back to Kiin Bio Weekly.
This week we’re going deep on a corner of drug discovery that rarely gets attention outside specialist circles: the computational tools being built specifically for peptides.
I came across Rodrigo’s review paper while researching the next primer in our series, and what struck me was how much progress has happened quietly in this field: new notation systems, fingerprinting methods, open-source toolkits, largely invisible to anyone not working directly in it. With peptide therapeutics drawing serious investment right now, it felt like exactly the right moment to bring it to a broader audience.
We got on a call, and the rest became this primer.
What’s your biggest time sink in early drug discovery process?
Peptides aren’t small molecules. They aren’t proteins. And for a long time, the computational tools didn’t know what to do with them.
For this primer, we spoke with Rodrigo Ochoa, a Senior Scientist at Novo Nordisk working on peptide design using AI, cheminformatics, and structure-based methodologies, and lead author of a new review paper on the open-source tools now available for peptide analysis.
Semaglutide did not become a blockbuster by accident. The GLP-1 receptor agonist behind Ozempic and Wegovy is a peptide - but not a natural one. Its half-life was extended from minutes to days through deliberate chemical modifications: a fatty acid chain, non-natural amino acid substitutions, structural tweaks that made it resistant to enzymatic degradation. The result was a molecule that could be dosed once a week and reach hundreds of millions of patients.
Designing the next one requires computational tools that barely existed a decade ago. A field called peptide cheminformatics is building them.
🧬 Why Peptides Need Their Own Computational Tools
The pharmaceutical industry has mature computational tools for small molecules. Biologics - proteins and antibodies - have their own tooling built around sequence analysis and structural biology. Peptides sit in between, and that middle ground has historically been underserved.
“Peptides are mixing nature from the small molecule world and also from the protein world,” Ochoa explains. “The challenge is basically to build tools that are customised for peptides, taking the best of both worlds.”
The scale of the problem compounds this. Medicinal chemists routinely work with non-canonical amino acids - hundreds of modified or synthetic building blocks beyond the standard 20 - that can dramatically alter a peptide’s properties. The resulting chemical space is so vast it cannot be explored broadly. In practice, scientists work within local regions: making targeted modifications around a known scaffold. That makes the quality of computational tools critical. Every prediction matters more when experiments are limited.
🧩 How Peptides Are Written Down, And Why It Matters
Before you can predict anything about a molecule, you need to represent it. For peptides, this has been the most fundamental bottleneck.
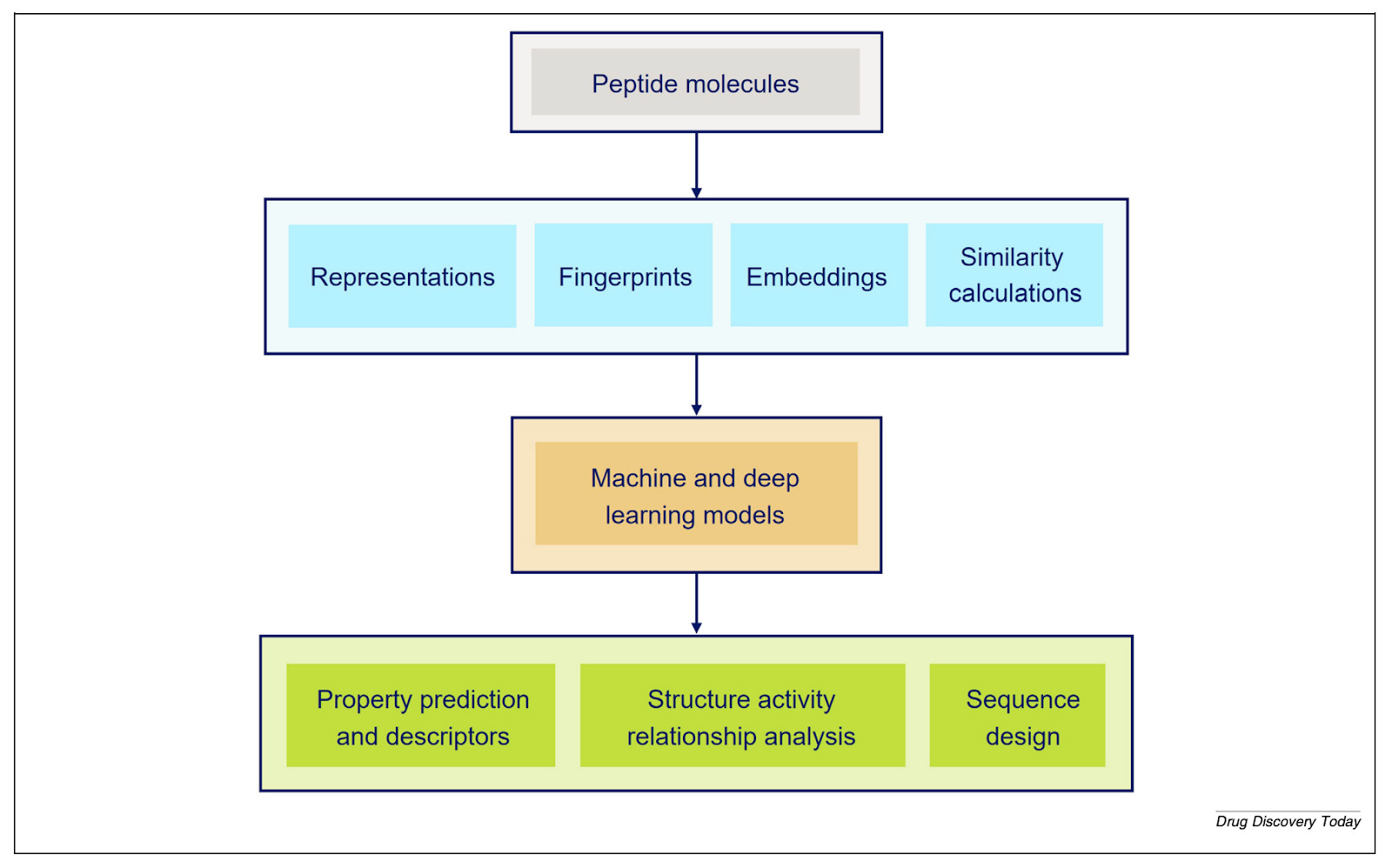
FASTA notation handles natural amino acids but cannot encode chemical modifications. SMILES can represent any chemical structure but loses the underlying sequence logic. Neither captures what peptide chemists actually need.
“The main issue is that we need a mix between a sequence-based representation and a full-atom representation,” Ochoa says. “Special notations like HELM or BILN rely on what we call a monomer dictionary - it contains the chemical information of each monomer and how each monomer can be connected to each other. That way we can make a full-atom representation of a whole peptide in a systematic way.”
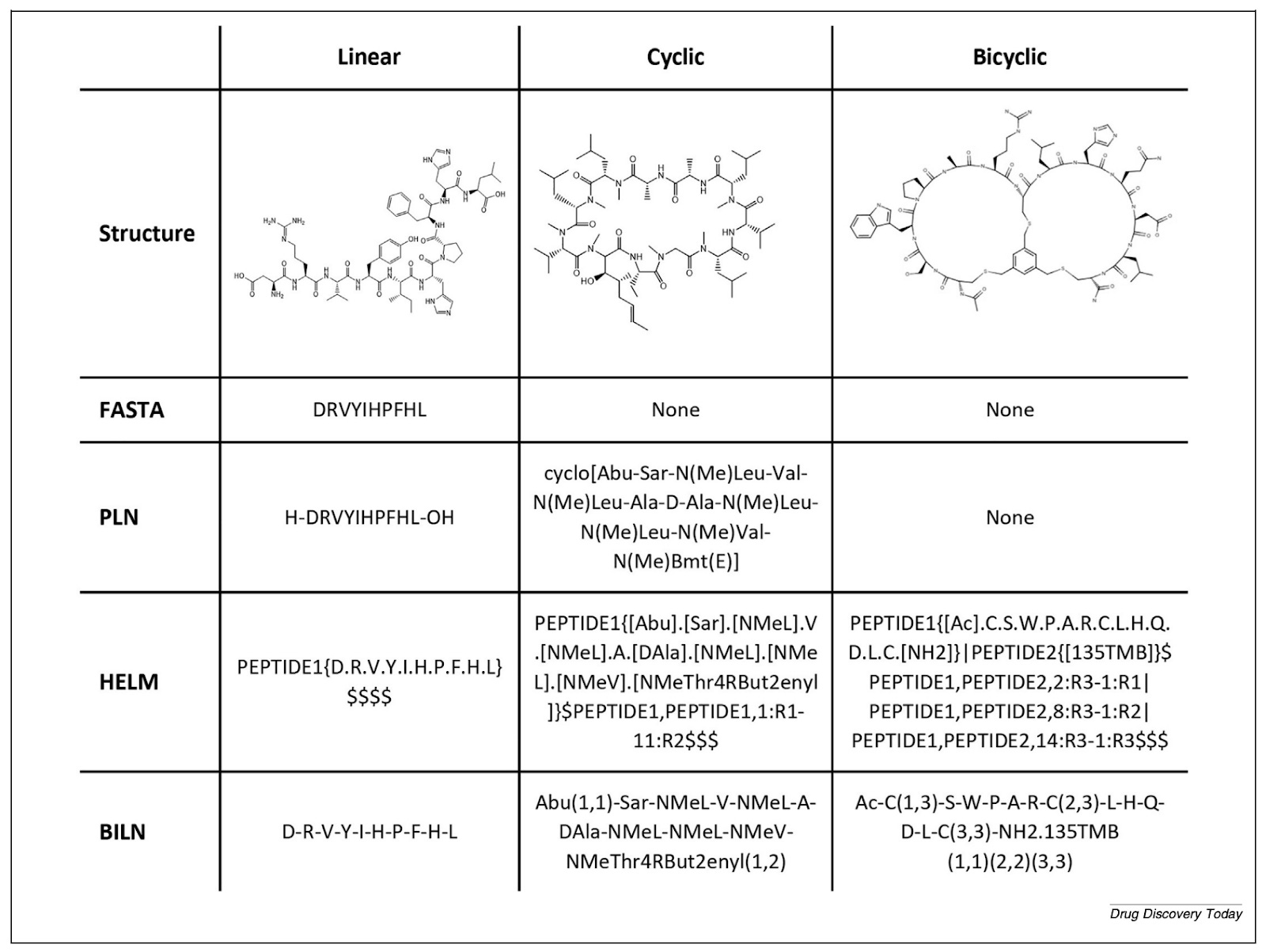
HELM notation, developed by the Pistoia Alliance in 2012, was the first major standardisation milestone. BILN, published in 2023 from work at Boehringer Ingelheim, took a more practitioner-friendly approach. “What we tried to do was make it as friendly as possible for the medicinal chemist,” Ochoa says - “something complex enough to include modified molecules, but that the chemist working with it can understand.” A Python package called pyPept now enables interconversion between notations, which matters when data arrives from multiple sources with different conventions.
🧮 From Fingerprints to Embeddings
Once you have a representation, you need to measure similarity - to ask whether a proposed modification is moving you toward or away from a desired property.
The standard approach for small molecules is Morgan fingerprints: fast, interpretable, and well-validated. But “you can lose some context about analogues or natural variants of the molecule,” Ochoa notes. The detection radius that works for small molecules doesn’t scale to the more complex architecture of peptides. Alternatives like MAP4 and monomer-based fingerprints address this by encoding similarity at the level of peptide building blocks rather than individual atoms.
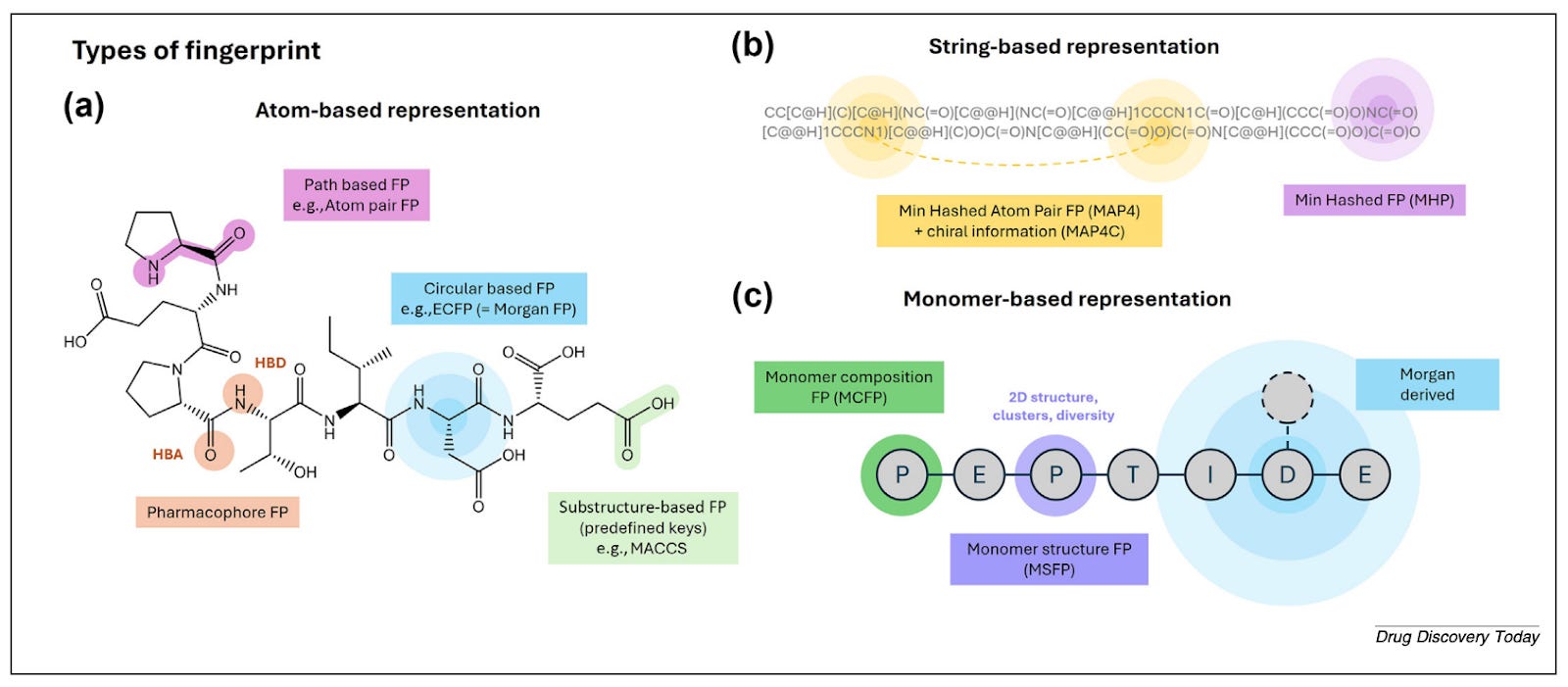
The more significant shift is toward embeddings. Protein language models like Meta’s ESM2 capture evolutionary and contextual information rather than explicitly encoded chemical structure.
“These embeddings are capturing evolutionary information,” Ochoa says. “What people are trying to do now is make embeddings that are more personalised for the peptide world - taking into account not only natural amino acids but also non-natural ones.”
In practice the two approaches complement each other. Fingerprints work well for simpler tasks like solubility prediction. Embeddings outperform on complex ones like binding affinity, where the relevant signal is harder to specify in advance.
⚙️ Peptide Cheminformatics Tools in Drug Discovery
Matched molecular pair analysis identifies which specific modifications statistically improve a given property. “The methodology lets you see how a small change in the chemical structure can have a big positive or negative impact on a potency readout or a physicochemical property,” Ochoa says. Clustering algorithms help design diverse compound libraries. Together, these tools support lead optimisation workflows from hit identification to refined candidates.
PepFuNN, an open-source toolkit from Novo Nordisk built on RDKit and biopython, is a direct example of industry contributing back to the community. “It’s a way for industry to contribute to the academic field,” Ochoa notes, “because at the end we also use the open-source methodologies available in the state of the art.” Web servers exist for researchers without programming skills - the value of these tools extends beyond computational specialists.
📊 Why Peptide Data Remains the Biggest Barrier
The field’s most underappreciated constraint is not methods - it is data. Public peptide datasets are scattered and inconsistently annotated. Commercial datasets are more standardised but confidential. ChEMBL is actively working to annotate peptide entries with HELM notation, but it remains ongoing work. Even excellent models are only as good as what they are trained on.
🔮 Generative AI and the Future of Peptide Design
Generative models are arriving. PepINVENT uses reinforcement learning to propose synthesisable peptides optimised for specific properties. The hallucination problem is real - models can generate chemically unrealistic structures - and knowing when a model is operating within its reliable range is critical.
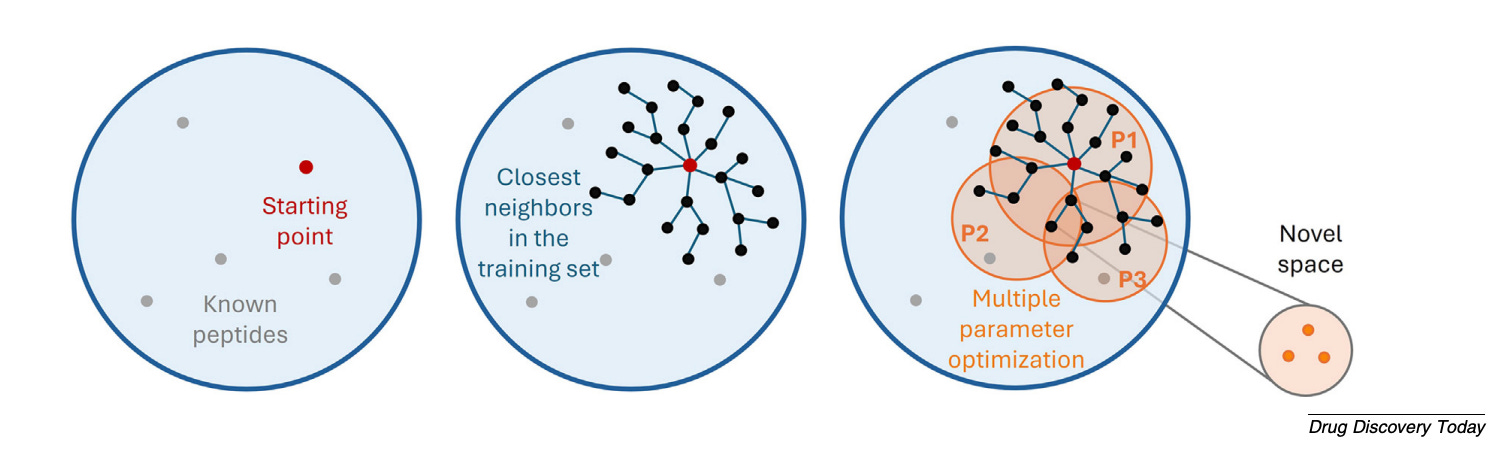
The near-term priorities: embeddings that properly capture non-canonical chemistry, generative models trained on better data, and LLM-based agents that lower the barrier for medicinal chemists without deep computational expertise. The direction is clear - from tools for specialists to tools for everyone who designs peptides.
Peptide therapeutics are drawing serious investment. Semaglutide is the headline, but behind it is a broader pipeline - GLP-1 analogues, cyclic peptides, stapled peptides, peptide-drug conjugates. The computational infrastructure is catching up. The data problem remains. But the foundations are solid enough to build on. Rodrigo’s paper is one of the first systematic attempts to map these tools in one place, and it arrives at exactly the right moment.
💬 Want to be featured in Kiin Bio Weekly?
Each issue we speak directly with researchers, scientists, and builders working at the frontier of AI in life sciences. If you're working on something in this space and think it would resonate with our community, I'd love to hear from you — fill out this form or reach out to me directly.
Found this useful? Forward it to a colleague in drug discovery or computational chemistry — it's the best way to help the newsletter grow.
Subscribe now to stay at the forefront of AI in Life Science. Every week: primers, deep dives, and direct conversations with the people building the field.
Connect With Us
Have questions on this or suggestions for our next deep dive? We’d love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website