
Absci’s Origin-1, JKU Linz’s MolecularIQ, and Genesis’s PEARL
Kiin Bio's Weekly Insights

Welcome back to your weekly dose of AI news for Life Science!
What’s your biggest time sink in the drug discovery process?
🧬 Origin-1: De Novo Antibody Design
What if AI could design therapeutic antibodies for targets we have never structurally seen before?
Most antibody discovery methods rely on precedent. Existing antibody–antigen structures, homologous complexes, or known epitope features. But what happens when none of that exists?
Origin-1, from the team at Absci, tackles this scenario directly: de novo antibody generation against zero-prior epitopes, regions with no structural precedent, no known binders, and limited sequence homology.
It is a full-stack generative platform, combining all-atom epitope-conditioned structure generation, paired CDR sequence design, and a custom co-folding scoring protocol to select designs predicted to bind with high specificity, developability, and atomic accuracy.
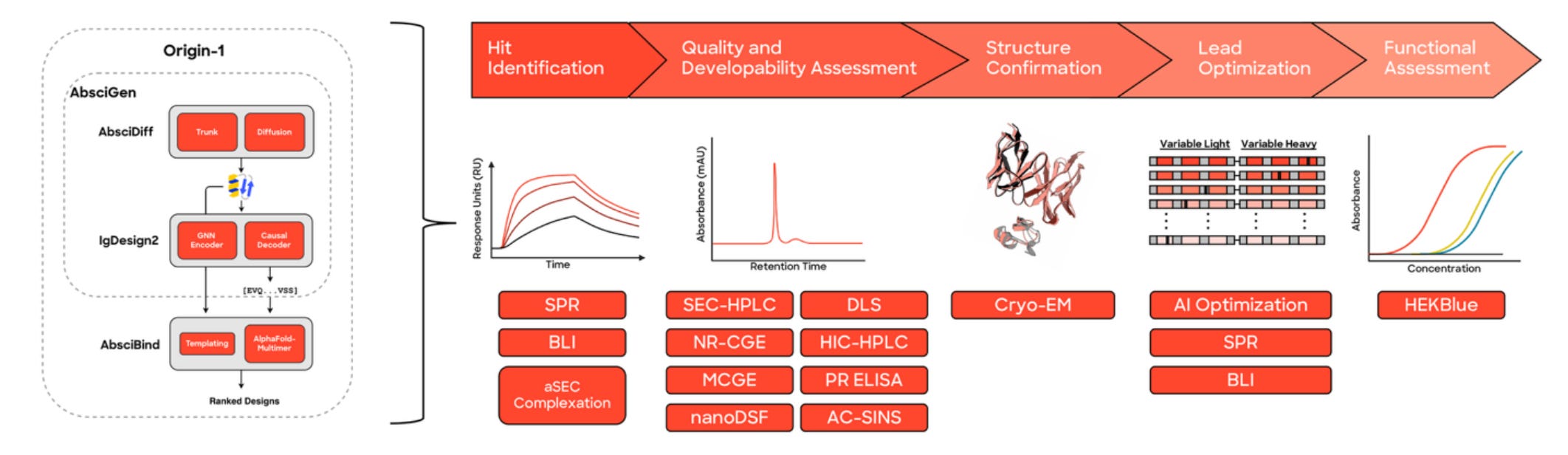
🔬 Applications and Insights
1️⃣ Antibody generation from scratch
For 10 structurally invisible targets (≤60 per cent sequence identity to anything with known complexes), Origin-1 generated fewer than 100 designs per target and identified high-confidence binders for 4: COL6A3, AZGP1, CHI3L2, and IL36RA.
2️⃣ Atomic-level structural accuracy
Cryo-EM revealed complexes at 3.0–3.1 Å resolution, matching computational models with DockQ scores of 0.73–0.83, showing strong agreement at atomic precision.
3️⃣ Functional activity and developability
For IL36RA, the de novo antibody acted as a functional antagonist and passed multiple biophysical and developability assays, showing real-world potential beyond the screen.
4️⃣ AI-guided affinity maturation
Starting from a fresh design, the team improved potency via in silico affinity maturation, ending with an antagonist at 104 nM IC50 with no human binder engineering required.
💡 Why It’s Cool
This is antibody generation without templates, scaffolds, or known contacts. Just the epitope, the physics, and the model. Origin-1 opens the door to programmable antibody discovery, even when nature has not left us a map.
📖 Read the paper
💻 Code
🧪 MolecularIQ: Testing Whether LLMs Understand Molecules
Can LLMs actually understand molecules, or are they just pattern-matching strings?
Many LLMs in chemistry claim strong performance on benchmarks like property prediction or synthesis planning. But what happens when you ask them to reason about molecular structure from first principles?
That is what MolecularIQ, from researchers at Johannes Kepler Universität Linz, is built to test. It is a benchmark of 5,111 symbolically verifiable tasks designed to probe whether models truly understand molecular structure beyond memorised patterns or training-set correlations.
It targets graph-level reasoning tasks like indexing, counting, ring membership, and multitasking across complex, randomised molecules.
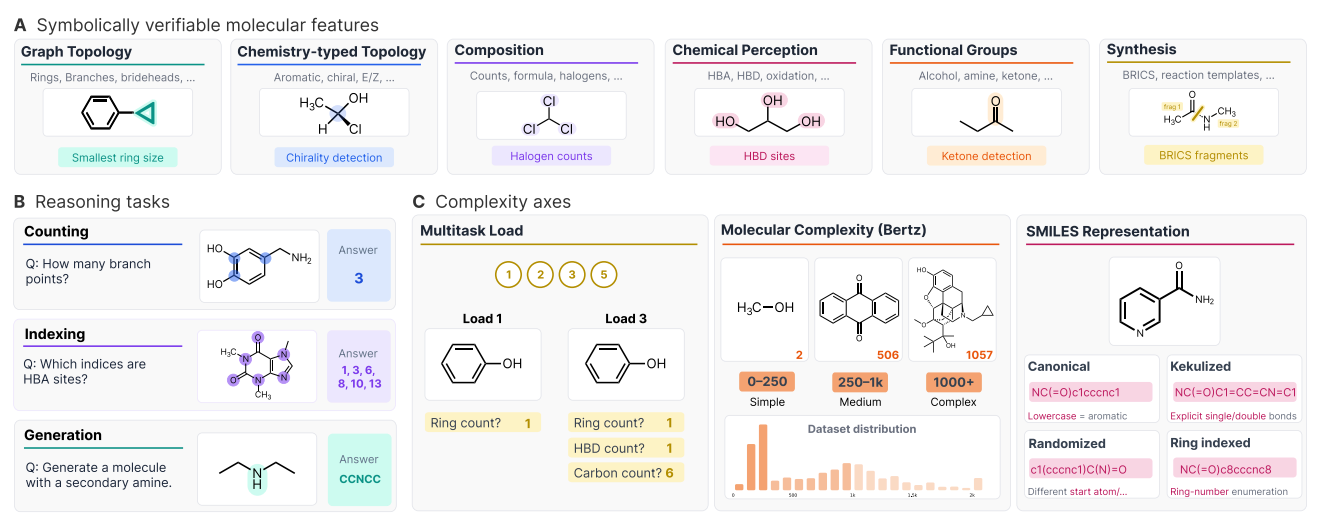
🔬 Applications and Insights
1️⃣ LLMs still struggle with core structural reasoning
The best model, GPT-OSS 120B, achieved 47.5 per cent accuracy. Chemistry-specialised models such as ChemDFM-R-14B peaked at 8.7 per cent. Most models failed to beat their own base versions.
2️⃣ Multitasking breaks models faster than complexity
Accuracy drops more when multiple questions are asked per molecule than when molecular size or structural complexity increases.
3️⃣ Non-canonical SMILES cause major failures
Models trained on canonical SMILES perform poorly on randomised or kekulised SMILES, revealing reliance on memorised string forms rather than true graph understanding.
4️⃣ Verbose answers are usually wrong
Incorrect responses tend to be longer, suggesting verbosity often masks confusion rather than understanding.
💡 Why It’s Cool
MolecularIQ is not just a scorecard. It shows where models fail, how they fail, and what reasoning they never learned. It is symbolically grounded, designed to avoid data leakage, and genuinely diagnostic for chemistry-capable language models.
📖 Read the paper
🧪 PEARL: Protein–Ligand Structure Prediction at Scale
Can we finally predict protein–ligand structures at scale with accuracy that actually holds up?
That is the promise of PEARL, Placing Every Atom in the Right Location, a foundation model for protein–ligand cofolding from researchers at Genesis Molecular AI.
Despite progress in deep learning for molecular structure, many models still run into familiar limits: not enough structural data, invalid poses, awkward architectures, and limited control at inference time. PEARL aims to combine precision, generalisation, and controllability in a single framework.
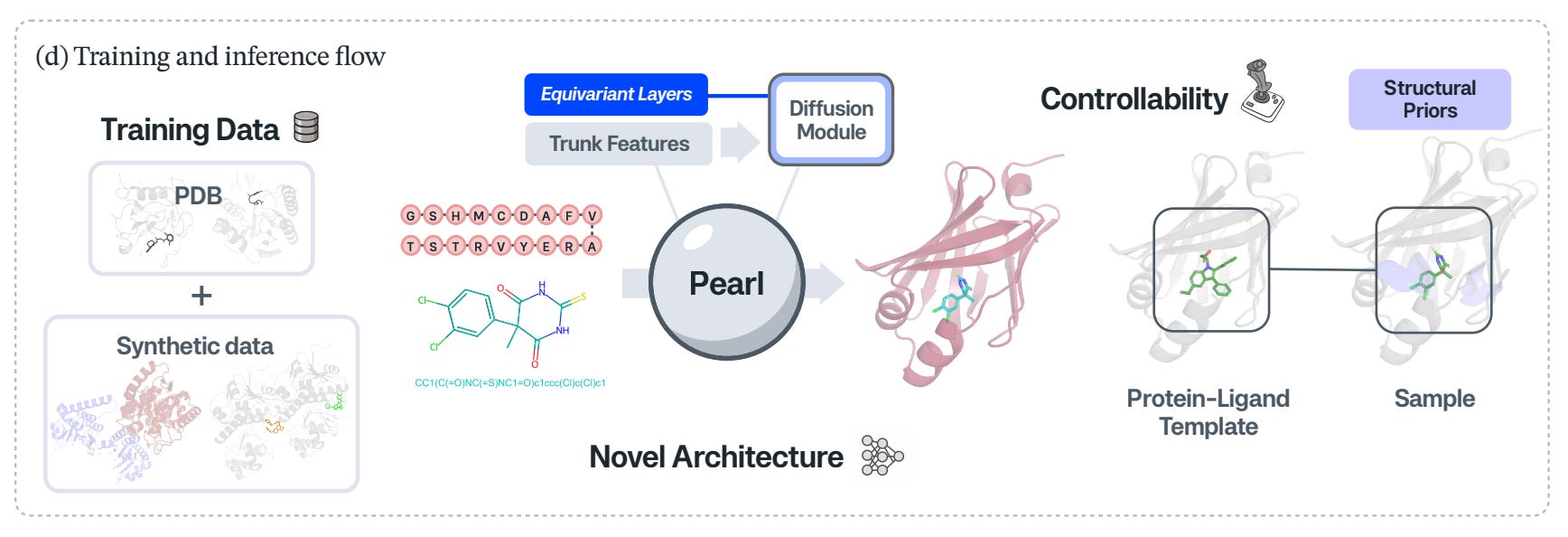
🔬 Applications and Insights
1️⃣ Large gains on real benchmarks
PEARL outperforms AlphaFold 3 and open-source baselines on Runs N’ Poses and PoseBusters, improving generation of physically valid <2 Å RMSD poses by 14.5 per cent and 14.2 per cent respectively.
2️⃣ Equivariant diffusion for stronger 3D reasoning
PEARL uses an SO(3)-equivariant diffusion module, building rotational symmetry into the model, which is critical for generalising across binding orientations.
3️⃣ Controllable inference with templating and conditioning
PEARL supports multi-chain templating, protein and small molecule inputs, and conditional or unconditional sampling, allowing users to guide generation without retraining.
4️⃣ Data scaling behaves predictably
Performance improves directly with the size of the synthetic training dataset, suggesting a clear path forward through data-centric scaling.
💡 Why It’s Cool
PEARL is not only more accurate, it is more usable. It folds, places, and refines atoms with symmetry-aware modelling and controlled generation, making structure prediction a programmable step in drug design workflows.
📖 Read the paper
💻 The code is not released yet unfortunately
Thanks for reading!
💬 Get involved
We’re always looking to grow our community. If you’d like to get involved, contribute ideas or share something you’re building, fill out this form or reach out to me directly.
Connect With Us
Have questions or suggestions? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website