
AI in Life Science: Weekly Insights
Weekly Insights | July 22, 2025

In this issue:
Welcome back to your weekly round-up of new tools and methods in life sciences research.
This week, we’re spotlighting three innovations helping researchers work more efficiently with single-cell data, bulk multi-omics, and protein interaction modelling:
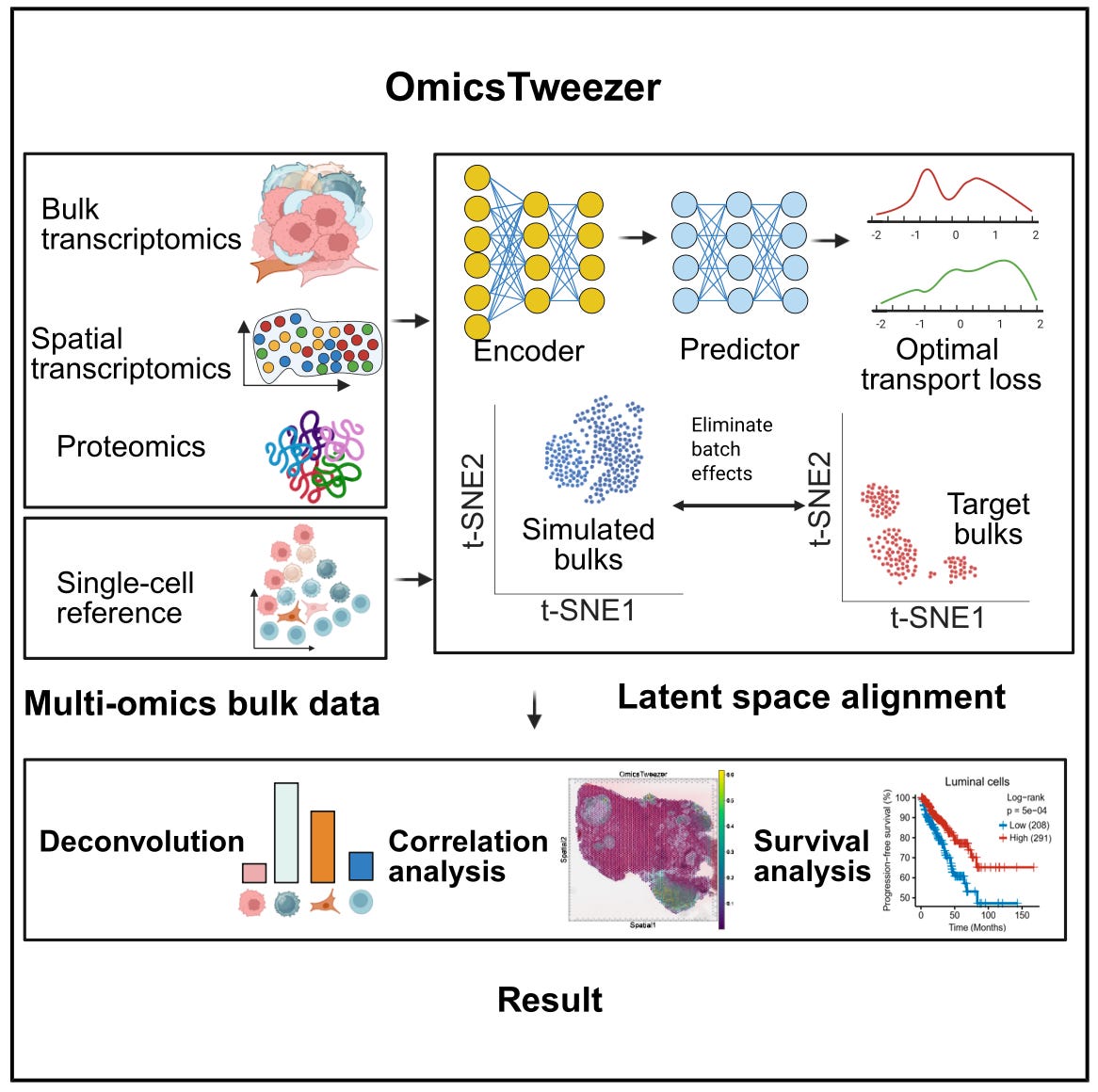
OmicsTweezer: Deep Learning for Multi-Omics Cell Type Deconvolution 🧫
GREMLN: Lightweight Foundation Models with Gene Regulatory Graph Priors 📊
ProtoBindDiff: Predicting Protein-Protein Binding with Interpretable Deep Learning 🧬
Explore how these approaches are improving resolution, efficiency, and insight across biological data analysis and molecular design.
What’s your most time consuming task along the drug discovery process?
We will send you open-source tools specific to your pain point.
OmicsTweezer: Deep Learning for Multi-Omics Cell Type Deconvolution 🧫
Bulk omics technologies offer broad and cost-effective profiling, but they obscure the diversity of cell types within complex tissues. OmicsTweezer provides a general solution for estimating cell type proportions from bulk data using single-cell references, even when measured on different platforms. The model combines a deep neural network with an optimal transport solver to align cross-modal distributions, correcting for batch effects and nonlinear feature shifts.
OmicsTweezer works across transcriptomics, spatial proteomics, and multi-modal inputs, making it highly applicable to cancer research, tissue profiling, and immune monitoring.
🔬 Applications
Tumour Composition Analysis - Identifies immune, stromal, and malignant cell populations in tumour biopsies using RNA or protein data from patient cohorts.
Cross-Modality Integration - Aligns single-cell references with bulk inputs collected using different protocols or technologies, including spatial and ATAC-seq data.
Population-Level Comparison - Quantifies shifts in cell type composition across treatment conditions, disease stages, or anatomical sites using standardised workflows.
📌 Key Insights
Benchmark Performance - Achieved Pearson correlation of 0.97 and RMSE of 0.06 on simulated bulk RNA mixtures, and greater than 0.94 correlation on real spatial proteomic samples.
Robust Across Platforms - Maintained accuracy within 2 percent of peak performance when reference and bulk data came from different tissues or measurement modalities.
Rare Cell Recovery - Detected immune cell subsets comprising less than 1 percent of tissue composition, outperforming CIBERSORTx and BayesPrism in sensitivity and specificity.
GREMLN: A Compact Foundation Model for Single-Cell Transcriptomics 📊
Single-cell foundation models can generalise across tasks, but often come with high computational costs and limited biological interpretability. GREMLN introduces a lightweight alternative: a 10 million parameter transformer that incorporates prior knowledge from gene regulatory networks. By combining structural graph priors with efficient model design, GREMLN improves accuracy on multiple single-cell benchmarks without the need for large-scale training.
The model supports common downstream tasks while remaining small enough to run on local infrastructure, making it accessible to labs with limited resources.
🔨Applications
Cell Type Classification - Accurately identifies distinct cell populations across tissue types and developmental states using fewer resources than large-scale models.
Differential Expression and Clustering - Produces biologically coherent cluster embeddings and detects subtle gene shifts across conditions or treatment groups.
Regulatory Network Integration - Incorporates transcription factor-target relationships into its learned representations, improving interpretability and reducing overfitting.
📌 Key Insights
Competitive Performance - Achieved F1 score of 0.92 for cell classification on the Human Cell Atlas benchmark, exceeding scGPT (0.86) and Geneformer (0.88) despite being 3 to 10 times smaller.
Computational Efficiency - Trained with 30 percent less GPU memory and converged up to 2.3 times faster than baseline models, making it suitable for desktop or local cluster environments.
Improved Biological Coherence - Increased recovery of known DE genes by 15 to 18 percent and produced clusters that preserved lineage relationships in hematopoietic and immune cell hierarchies.

ProtoBindDiff: Ranking Protein Binding Differences Without Templates 🧬
Understanding how structural changes or point mutations affect protein–protein binding is critical for therapeutic antibody design and rational protein engineering. Most existing tools rely on co-crystal structures, docking, or energy functions that limit their applicability to known complexes. ProtoBindDiff offers a general framework for predicting relative binding differences directly from structure, without requiring co-complex inputs or template-based modeling.
By training on paired examples and using attention-based encoders, the model captures binding strength shifts across different protein families. Its performance is validated on real benchmarks and its outputs include interpretable residue-level scores that help guide design.
🔨Applications
Mutational Scanning - Enables rapid in silico screening of point mutations across antigen–antibody, receptor–ligand, or cytokine–receptor interfaces to prioritise candidates for experimental validation.
Therapeutic Engineering - Supports affinity maturation, paratope optimisation, and epitope targeting by comparing sequence variants of antibodies or binding proteins.
Structure-Free Ranking - Functions without pre-aligned templates or docking, making it suitable for exploring novel targets with only structural monomers or unpaired complexes.
📌Key Insights
High Predictive Accuracy - Achieved a Spearman correlation of 0.79 and RMSE of 0.83 kcal/mol on SKEMPI v2.0, outperforming STRUM and MutaBind2 on the same benchmark.
Generalisation to Novel Targets - Retained greater than 0.75 correlation on complexes without homologs in the training set, showing it learns transferable features across protein families.
Mechanistic Interpretability - Attention scores aligned with known binding interfaces in over 85 percent of test cases, offering residue-level insight into changes in affinity.
Did you find this newsletter insightful? Share it with a colleague!
Subscribe Now to stay at the forefront of AI in Life Science.
Connect With Us
Have questions or suggestions? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website