
AI in Life Science: Weekly Insights
Weekly Insights | January 22, 2024

In this issue:
Welcome back to your weekly dose of AI news for Life Science!
This week, we have some exciting new models lined up for you:
PPDock: Pocket Prediction-Based Protein–Ligand Blind Docking🔬
PLMFit : Benchmarking Transfer Learning with Protein Language Models for Protein Engineering 💿
Dive into these game-changing innovations and explore how they are transforming the biotech and healthcare landscapes!
PPDock: Pocket Prediction-Based Protein–Ligand Blind Docking🔬
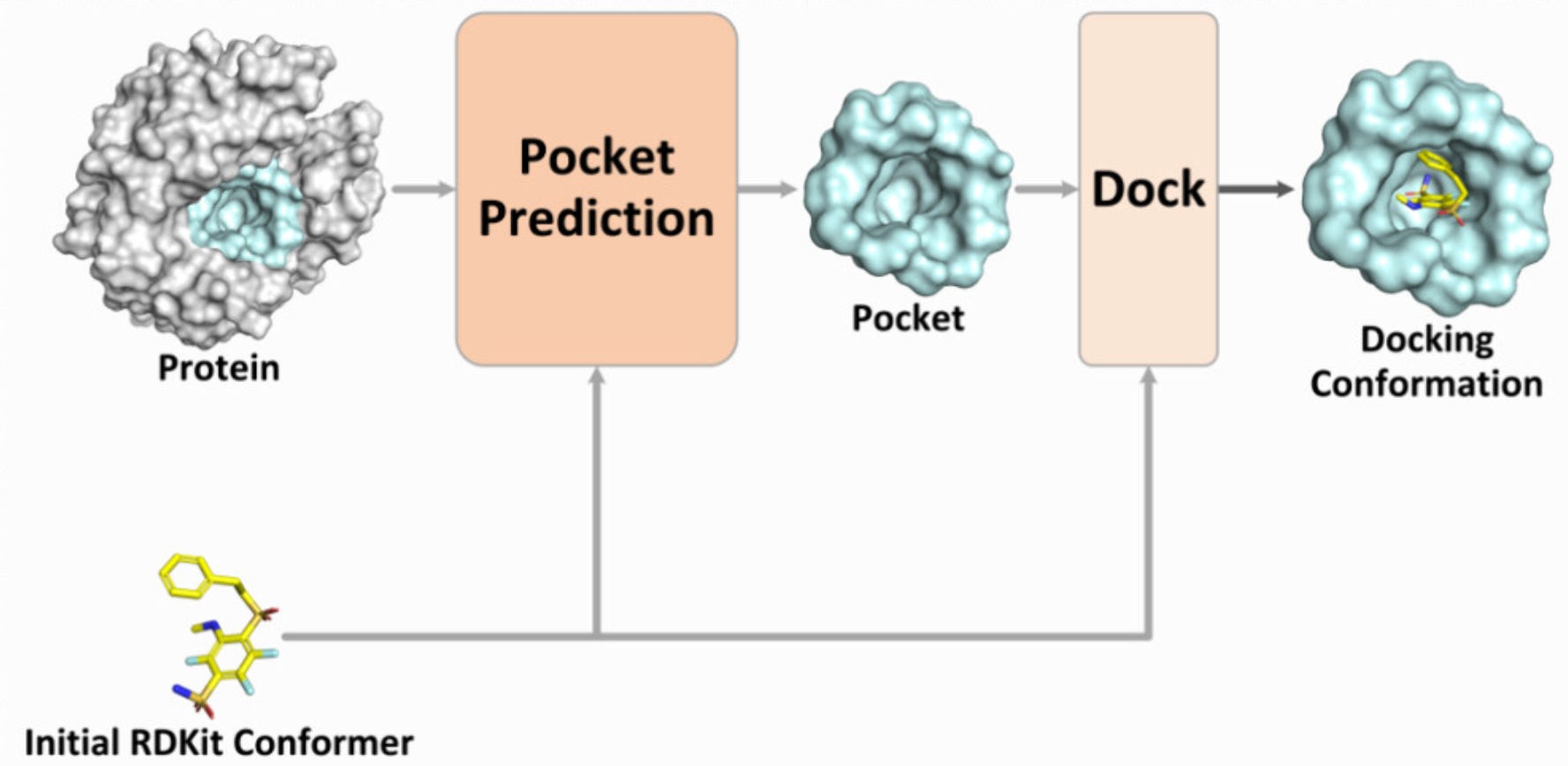
The new year started strong with new protein design tools! Predicting the docking conformation of a ligand in the protein binding site (pocket), i.e., protein–ligand docking, is crucial for drug discovery. Traditional docking methods have a long inference time and low accuracy in blind docking (when the pocket is unknown). Recently, blind docking techniques based on deep learning have significantly improved inference efficiency and achieved good docking results. However, these methods often use the entire protein for docking, which makes it difficult to identify the correct pocket and results in poor generalisation performance. Introducing PPDock, a two-stage docking paradigm, where pocket prediction is followed by pocket-based docking.
📌 Key Insights:
PPDock outperforms existing methods (e.g. DiffDock) in nearly all evaluation metrics, demonstrating strong docking accuracy, generalisation ability, and efficiency (36.4% of predictions with RMSD below 2 Å)
PPDock runs extremely fast, with the inference as low as 0.06 s per ligand, significantly faster than DiffDock that takes 5-10 seconds!
PPDock was trained on PDBbind v2020 which contain complex structures with ligand and its complete protein receptor. From PDBbind, 17,299 complex structures are used for training, 968 for validation, and 363 for testing.
PLMFit : Benchmarking Transfer Learning with Protein Language Models for Protein Engineering 💿
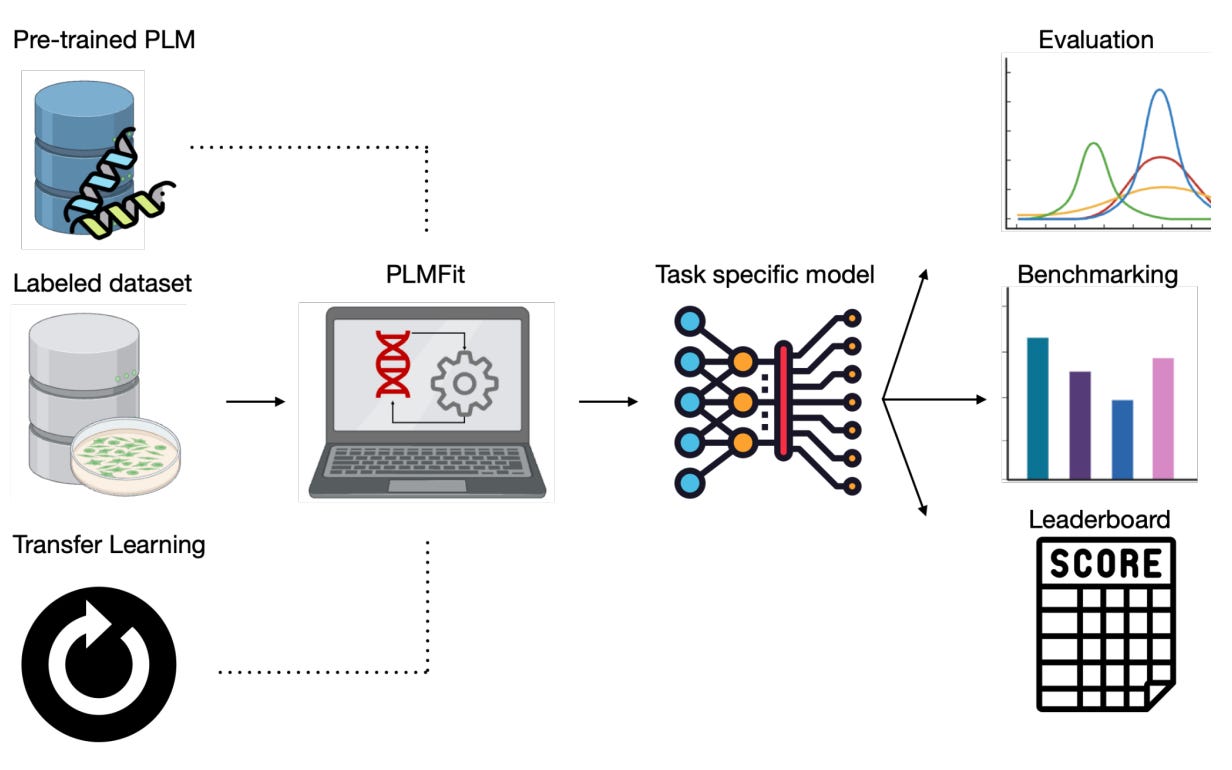
In the spirit of Large Language Models (LLMs) such as ChatGPT or Claude, the same technology is more and more used for solving protein engineering applications, known as Protein Language Models (PLMs). The most well-known example in this family is AlphaFold, with its founders decorated with a Nobel Prize in 2024. However, building such a model from scratch is rapidly very expensive, whereas researchers often resort to Transfer Learning, an efficient technique that drastically decreases the cost of training. Easy on paper, but harder to put in place, PLMFit is presented here as a tool that aims at democratising this process, with varying levels of computational expertise. It allows fine-tuning of any state-of-the-art model without requiring in-depth programming knowledge.
📌 Key Insights:
Open-source GitHub project for fine-tuning any state-of-the-art PLM without requiring in-depth programming knowledge.
Testing on three state-of-the-art models (ESM2, ProGen2, ProteinBert) with different Transfer Learning approaches and applied to various protein engineering datasets.
Rationalize with these tests the understanding behind efficiently fine-tuning a PLM with Transfer Learning.
molli: A General Purpose Python Toolkit for Combinatorial Small Molecule Library Generation, Manipulation, and Feature Extraction 💻
Manipulating in silico molecular libraries is one of the main tasks of cheminformaticians, but it is also one of the most challenging due to the lack of standardization among the most commonly used tools. In this context, let’s introduce the MOLecular LIibrary toolkit (“molli”), which is a Python-based cheminformatics module that provides easier access to many computational tools. From the Python RDKit package to the ORCA and xTB software for quantum computations, molli offers an API for molecule manipulations, combinatorial library generation with stereochemical fidelity from plain CDXML files, as well as a parallel computing interface.
📌 Key Insights:
Provide a full representation of molecular graphs, geometries, and geometry ensembles with no implicit atoms.
Allow interfacing with widely used computational chemistry programs such as OpenBabel, RDKit, ORCA, and xTB/CREST.
Benchmarks have been made on various hardware platforms, and common workflows are demonstrated for different tasks ranging from optimized grid-based descriptor calculation on catalyst libraries to NMR chemical shift prediction workflows from CDXML files.
Did you find this newsletter insightful? Share it with a colleague!
Subscribe Now to stay at the forefront of AI in Life Science.
Connect With Us
Have questions or suggestions? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website
Great read! I'm particularly impressed by PPDock's two stage approach and how it contributed to its performance and speed. Hope it achieves the same results in real-world projects!