
AMR: Generative Antibiotic Design
Deep Dive | Edition 2

Welcome back to the deep dive, where we break down the AI tools and data reshaping how new drugs are discovered. In each edition, we speak directly with the teams behind these tools to explain what they solve, how they work and where they are going next.
This time, we spoke with Aarti Krishnan about her MIT team’s project that pushes generative AI beyond vast enumerated libraries and into true de novo antibiotic design.
Antimicrobial resistance (AMR) is one of the hardest challenges in medicine. Infections like gonorrhea and staph are already resistant to most of the drugs we have, and industry pipelines are drying up. For decades, computational discovery has relied on screening massive libraries made up of billions of molecules that still cover only a sliver of possible chemical space and the main hurdles are compute power and lack of novelty.
De novo design addresses this directly: while synthesisability becomes the bottleneck, any molecules that can be made are especially valuable since their novelty offers a real chance to outpace resistance.
🔴 The Problem
So, antibiotic discovery is well known to be in crisis. Pathogens such as Neisseria gonorrhoeae and Staphylococcus aureus are now listed by the CDC as “urgent” and “serious” threats, with resistance emerging to every single frontline drug. Yet between 1980 and 2003, only five new antibiotics were launched by the top 15 pharma companies. The economics are clearly broken and evolutionary pressure makes the bacteria rapidly adaptable to new drugs.
Traditional computational chemistry discovery pipelines lean heavily on enumerated chemical libraries like Enamine that max out at around 1011 molecules. But the space of possible drug-like molecules is closer to 1060. That means most of chemical space, and most of the structural novelty needed to beat resistance, is ultimately unexplored. Even when promising hits are found in these libraries, medicinal chemists run into the next bottleneck: synthesizability. Molecules that look great in silico often can’t be made in the lab, stalling projects before they even begin.
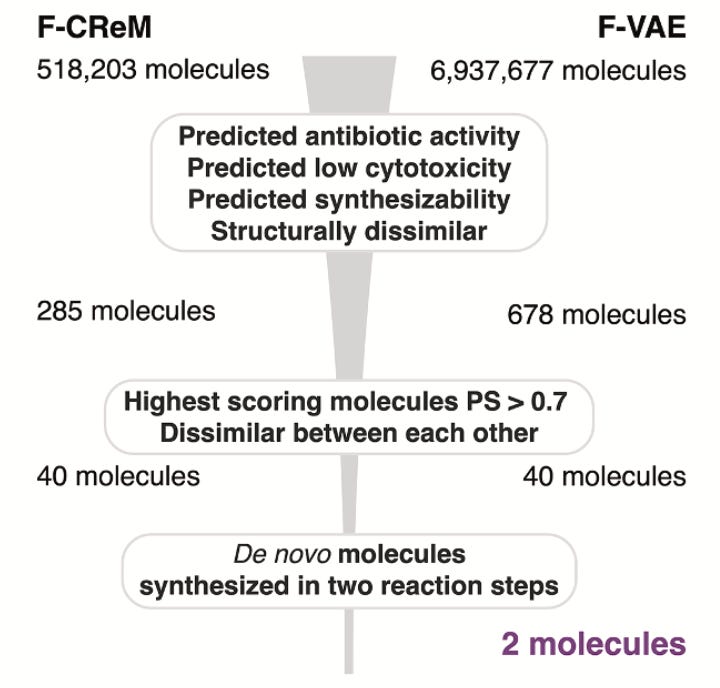
Aarti and her team saw this problem first-hand. For their fragment-based approach to de novo antibiotic design, they started with over 7 million generated molecules and filtered down to 120 with high antibacterial scores. When chemists attempted synthesis, 90% proved intractable. In the end, just two molecules were made. At that point, most teams might have called it a failure. What were the odds that either would turn out antibacterial? Surprisingly, one of those molecules turned out to be highly potent and highly selective against pathogenic gonorrhea and not commensal Neisseria bacteria. Eureka! That narrow-spectrum activity quickly flipped this story from disappointment to breakthrough.
💡 The Idea
Their pipeline combines graph neural networks (Chemprop) to score antibacterial activity, with genetic algorithms (CReM) and variational autoencoders (VAEs) to generate new molecules. But unlike many generative pipelines, synthesisability wasn’t an afterthought, it was used as the prime starting point. Every candidate was filtered through tools like SPARROW and ASKCOS (2.0 version just launched), which predict retrosynthetic routes, reagent availability, and cost. Without these filters, most of the molecules wouldn’t stand a chance in the lab.
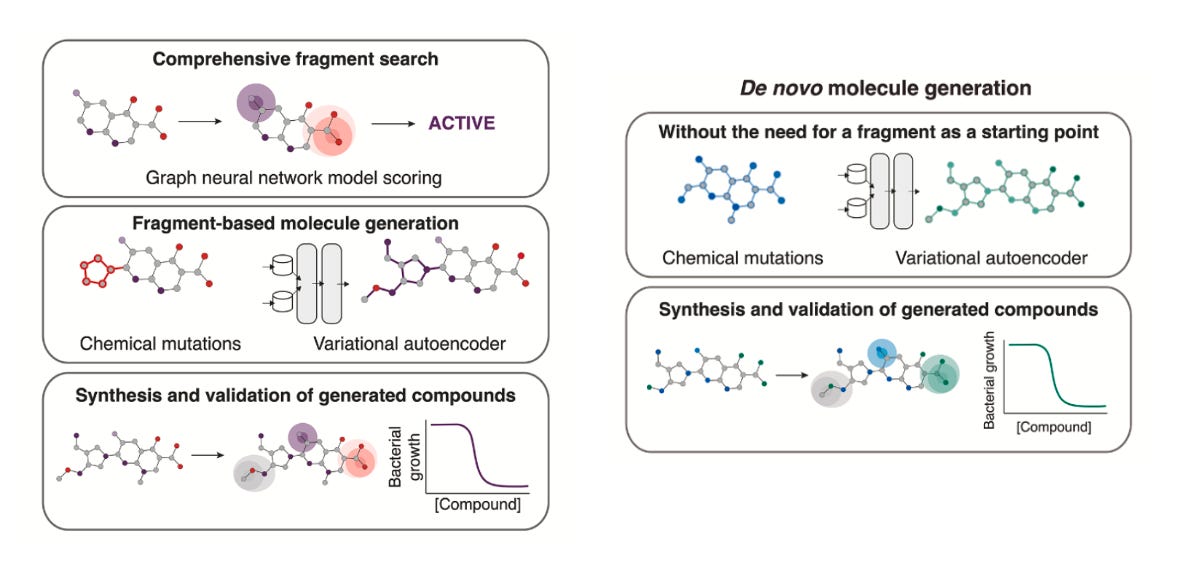
Two complementary strategies emerged:
Fragment-based expansion: starting with promising chemical fragments, then growing them into full molecules.
Why this approach? This is where the team began. By anchoring generation around real, drug-like fragments, they increased the odds of generating something effective and synthetically tractable. That led to NG1, a novel, narrow-spectrum antibiotic that kills pathogenic gonorrhea but spares commensal species. It worked in a mouse infection model and showed a membrane-related mechanism of action, possibly through the lipooligosaccharide export protein LptA, a long-proposed but undrugged target.Unconstrained de novo generation: starting from nothing (or methane, water or ammonia) and allowing the models to create entirely new scaffolds.
Why this approach? Once confident in their filters, the team explored a more open-ended strategy. This route produced DN1, active against Staphylococcus aureus, including MRSA. While they couldn’t pinpoint a single protein target, they observed significant membrane damage and most notably, no evolutionary resistance emerged which is a rare and highly valuable property.
From these approaches, the team synthesised 24 candidates. Seven had antibacterial activity and these two, NG1 and DN1, stuck out like exciting sore thumbs.
The takeaway? While the project began with fragment-based design, it ultimately didn’t matter where you started. Both strategies, with and without fragments, delivered promising leads, as long as synthetic accessibility was considered.
🔬 Why It’s Different
Plenty of teams have leveraged generative models for drug design. What sets this one apart for us is the integration of AI creativity with real-world chemistry. The marrying of biology and chemistry in the early stages which stands to the model later in its work.
True de novo design: Instead of just screening what’s already in very large libraries such as Enamine or ZINC, the models generate previously unenumerated scaffolds, opening up regions of chemical space no one has touched.
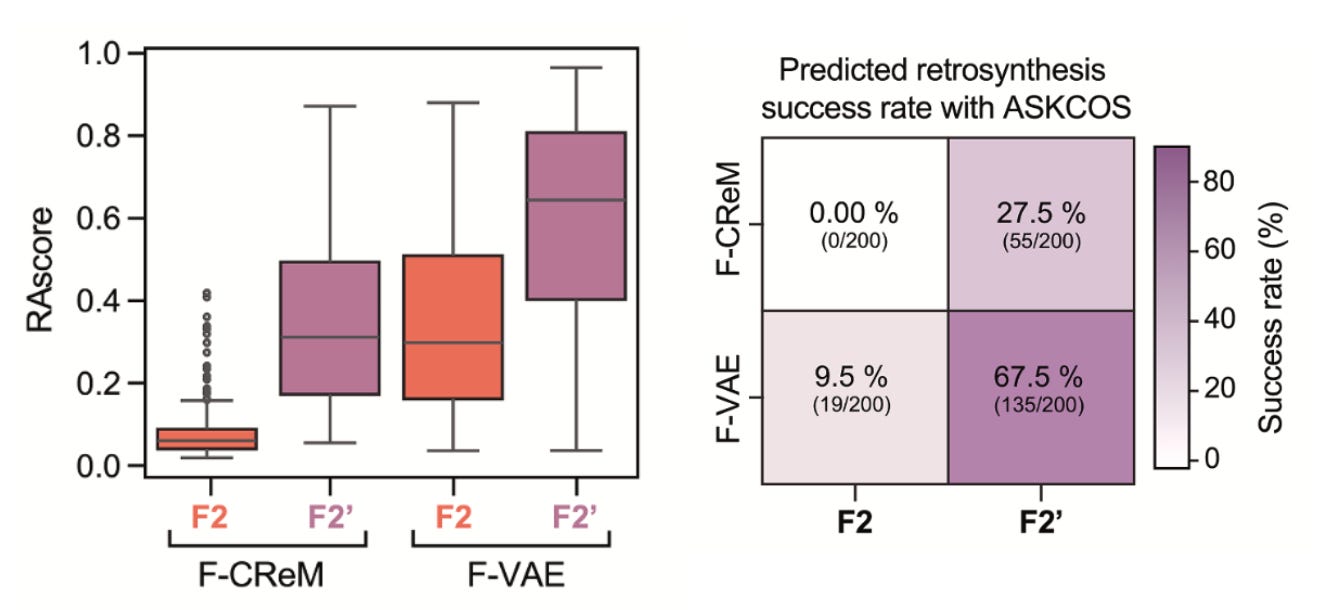
Synthesisability-first: Most generative pipelines fail at the lab bench. Here, every candidate is filtered through retrosynthesis planners like ASKCOS before chemists ever pick up a flask. That kept the focus on molecules that could actually be made.
Phenotypic anchoring: By training against whole-cell activity rather than predefined targets, the models are free to discover molecules with new modes of action, like NG1, which zeroed in on a previously undrugged lipooligosaccharide export protein.
Striking hit rates: Conventional high-throughput screens often yield hit rates well under 1%. In contrast, this de novo pipeline delivered 7 actives from 24 synthesised compounds (29%), which is clearly an enormous jump in efficiency.
The bottom line is the result isn’t just more molecules, its new structural classes of antibiotics with in vivo efficacy, against two of the most resistant pathogens we face today.
🔮 The Future
PhareBio, a Cambridge-based non-profit organisation, is advancing several candidate molecules, such as NG1 and DN1, toward optimisation and preclinical development. These efforts include designing and generating analogues to map structure-activity relationships and conducting early ADME and toxicity studies to evaluate pharmacological properties and safety. Promising molecules will then be formulated and tested in mouse models of systemic infection, laying the groundwork for preclinical trials.
Building on the success of their pipeline, Aarti and her colleagues at MIT plan to extend their work to other high-priority pathogens such as Pseudomonas aeruginosa, Klebsiella pneumoniae, and drug-resistant Mycobacterium tuberculosis, broadening the potential impact against critical antimicrobial resistance threats. The team is also developing deep learning platforms to streamline hit-to-lead optimisation, aiming to dramatically reduce development timelines.
📄 Read the paper!
⚙️ Try the code out.
👨🔬 Get in touch with Aarti.
Thanks for reading!
Did you find this newsletter insightful? Share it with a colleague!
Subscribe Now to stay at the forefront of AI in Life Science and keep up with this upcoming season of deep dives.
Connect With Us
Have questions or suggestions for our next deep dive? We’d love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website