
Biohub's ESM, Georgia Tech's SynFit, and UCSF's OpenADMET
Kiin Bio's Weekly Insights

Welcome back to your weekly dose of AI news for Life Science!
I keep coming back to this question of whether the protein ML field has a modelling problem or a data problem. This week's papers make the case from both sides. Biohub's ESM release argues that scale and architecture can solve binder design from scratch. OpenADMET argues the opposite: that no model will work until the training data stops being inconsistent rubbish. SynFit sits somewhere in between, showing what you can get when you have good multi-property data and a framework that knows how to use it.
We just launched our Kiin Pioneer Programme, giving academic and nonprofit research teams one year of free access to our drug discovery platform!
KiinOS is a platform where scientists can run literature reviews, target discovery, and bioinformatics in one place. It keeps a record of what’s been done, by who, and what came out of it. So if one person finds a promising target and someone else has relevant data, the platform connects those results and suggests what to pursue next.
We’re looking for teams asking: which targets should we prioritise? How do we interpret conflicting evidence? Which hypotheses are worth testing next?
There’s no cost, no data transfer, and all IP stays with your institution. Applications close in August, with the first cohort starting September.

ESM: A World Model of Protein Biology
🔬 We can predict how proteins fold, but designing new ones that actually bind specific targets, particularly antibodies, still requires months of expensive lab screening just to find starting candidates.
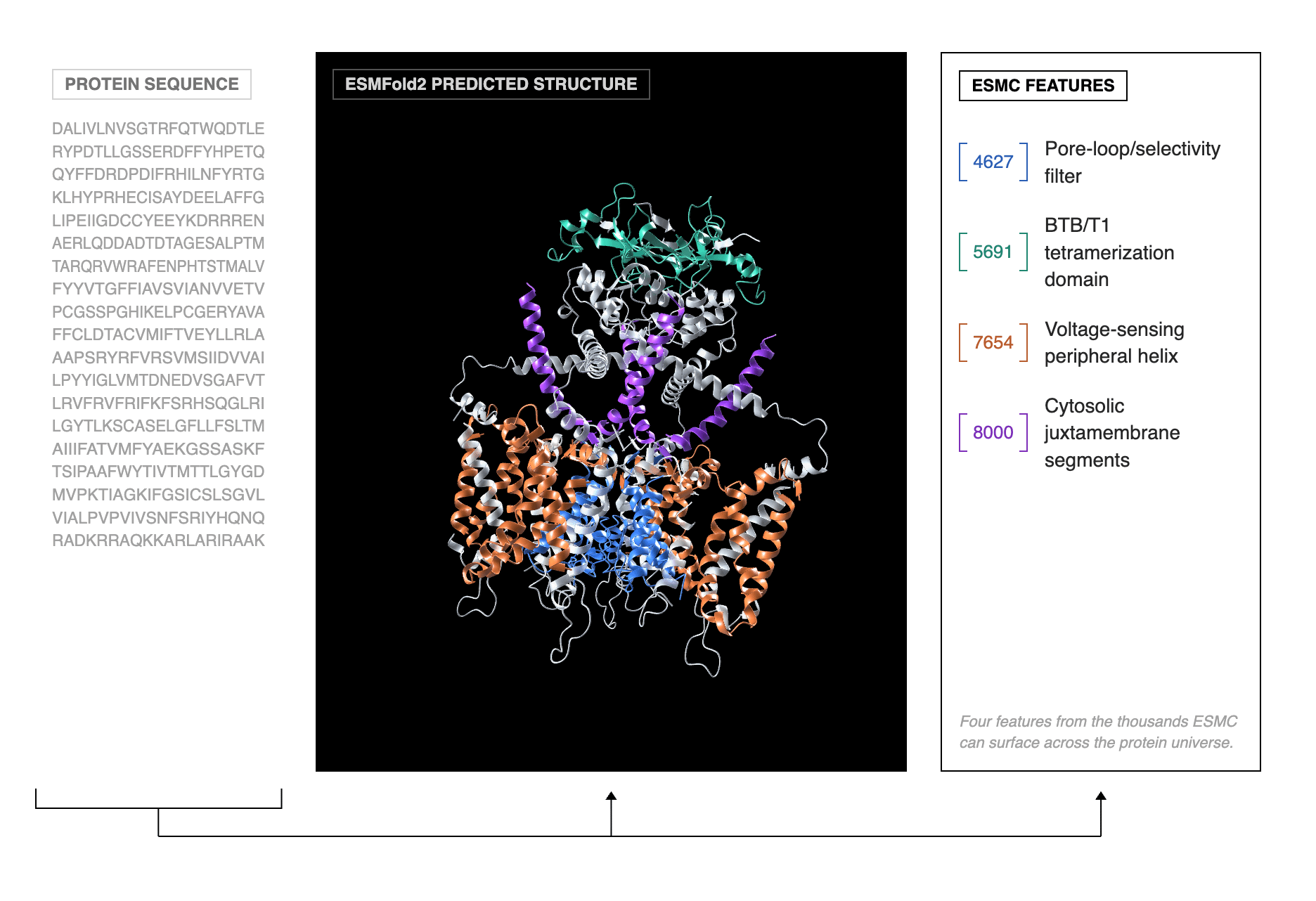
Biohub has released ESM: a protein language model (ESMC, trained on 2.8 billion sequences), a structure prediction and design model (ESMFold2), and a map of 6.8 billion sequences (ESM Atlas). All MIT-licensed.
🧬 ESMFold2 learns protein representations from evolutionary data, then searches that learned space for proteins predicted to bind a given target. It scores candidates using its own confidence estimates, so the entire design loop is computational. Structure prediction runs from a single sequence without needing alignment databases.
⚡ Designed binders for five cancer/immunology targets were validated in the lab: minibinder success rates of 70%, scFv antibody success rates of 21%. A PD-L1 binder hit 4.3 nM affinity and blocked immune checkpoint suppression in cells. Cryo-EM confirmed an EGFR binder matched the prediction at 1.2 Å RMSD.
🧪 Where This Fits
This positions ESM as a competitor to both AlphaFold 3 (for structure prediction) and RFdiffusion/ProteinMPNN (for binder design). The antibody-antigen prediction results are interesting because this is where AlphaFold has historically struggled most. The binder design piece is where Biohub is making their boldest claim: that the earliest stage of therapeutic protein discovery can happen computationally in days rather than months. Worth noting that the 21% scFv success rate, while a step up from near-zero for most computational methods, still means roughly 80% of designs fail in the lab. These are first-round hits. David Baker’s group (RFdiffusion) and Generate Biomedicines (Chroma) are the obvious comparisons for generative protein design. ESMFold2 is differentiated by working from a language model backbone rather than a diffusion architecture, which changes how it scales with compute at inference time.
💡 Why This Is Cool
The sparse autoencoder analysis is what I find most thought-provoking. ESMC independently recovered biological concepts like the nucleophilic elbow motif across 75 of 99 relevant enzymes, despite never being told what one is. That’s a model learning the grammar of protein biology from sequence alone. Whether the binder design numbers hold up across a broader and more difficult target set remains to be seen. The cryo-EM validation is reassuring, but five targets is still five targets.
📄 Read their press release.
💻 Try the tool.
SynFit: Synergistic Contrastive Learning for Multi-Objective Protein Fitness Prediction and Optimisation
🔬 Protein engineering almost always requires optimising multiple properties at once, but current ML fitness predictors handle each property independently. Train separate models for yield and selectivity and you’ll get variants that excel at one while tanking the other.
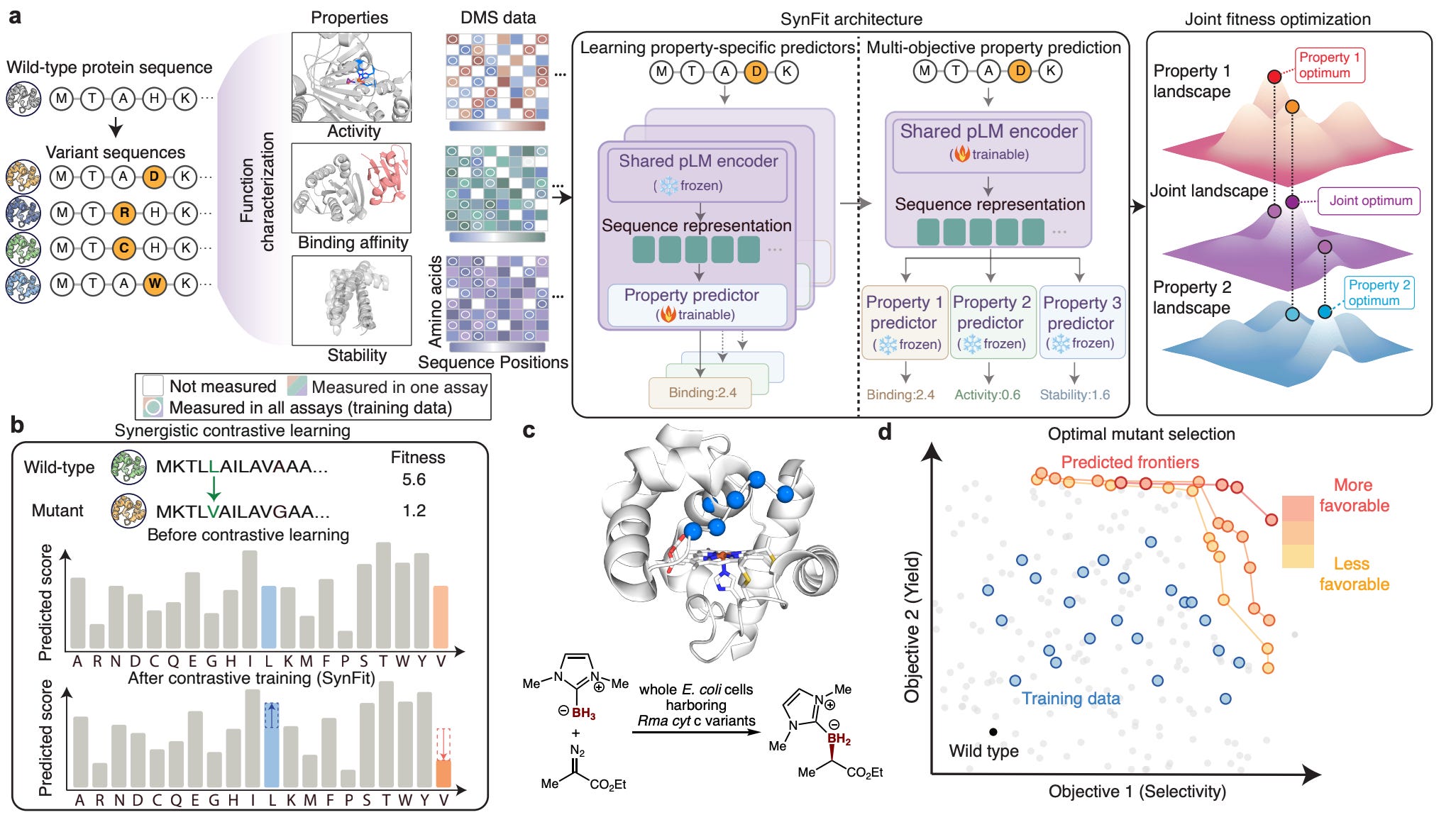
Georgia Tech and UC Santa Barbara developed SynFit, a multi-objective framework that fine-tunes protein language models on experimental fitness data across multiple assays simultaneously.
🧬 SynFit combines a shared ESM2 encoder with property-specific prediction heads, using contrastive learning to capture cross-property relationships from deep mutational scanning data. Predictions are integrated via Pareto sorting to find variants that improve everything at once.
⚡ On Pareto front analysis across 20 proteins, SynFit hits the optimal front 70% of the time versus 60% for ProteinNPT and 55% for ConFit. The wet-lab result is more convincing: 83 out of 100 designed hextuple mutants for a biocatalytic borylation enzyme showed simultaneously improved yield and enantioselectivity, with multiple variants beating everything in the training data.
🧪 Where This Fits
This sits squarely in the protein engineering workflow after you have initial DMS data and want to explore combinatorial sequence space. ConFit, from the same group, is the direct predecessor, and SynFit extends it with the multi-objective training component. ProteinNPT (Notin et al., ICML 2023) tackles a related problem through non-parametric transformers but doesn’t explicitly model cross-property correlations. The wet-lab validation on biocatalytic borylation is well chosen because it’s a genuinely new-to-nature reaction where directed evolution data is sparse, making the Pareto optimisation problem harder. The limitation is that you still need initial multi-property DMS data for your protein of interest, which isn’t always available. The KRAS case study (identifying shared functional residues across six binding partners) is a nice mechanistic demonstration but relies on an unusually well-characterised system.
💡 Why This Is Cool
The 83/100 result is more impressive than any of the benchmark numbers. Getting computationally designed hextuple mutants to simultaneously beat the training set on both yield and enantioselectivity, in a single round without iterative screening, is a practical result that enzyme engineers will care about. It suggests ML-guided combinatorial design can start to compress the “design-build-test” cycle for multi-objective problems. The architecture is straightforward enough that adoption shouldn’t be difficult once the code is released.
📄 Read the paper.
💻Try the code.
Mapping the Avoid-ome: A Systematic Open-Science Approach to Predictive ADMET
🔬 Around 30% of clinical drug failures trace back to ADMET problems. The ~100 proteins responsible (CYPs, hERG, transporters, nuclear receptors) are well known, but existing ML models train on data cobbled from dozens of labs using different protocols. A recent analysis found almost no correlation between IC50 values for the same compound measured by different groups.
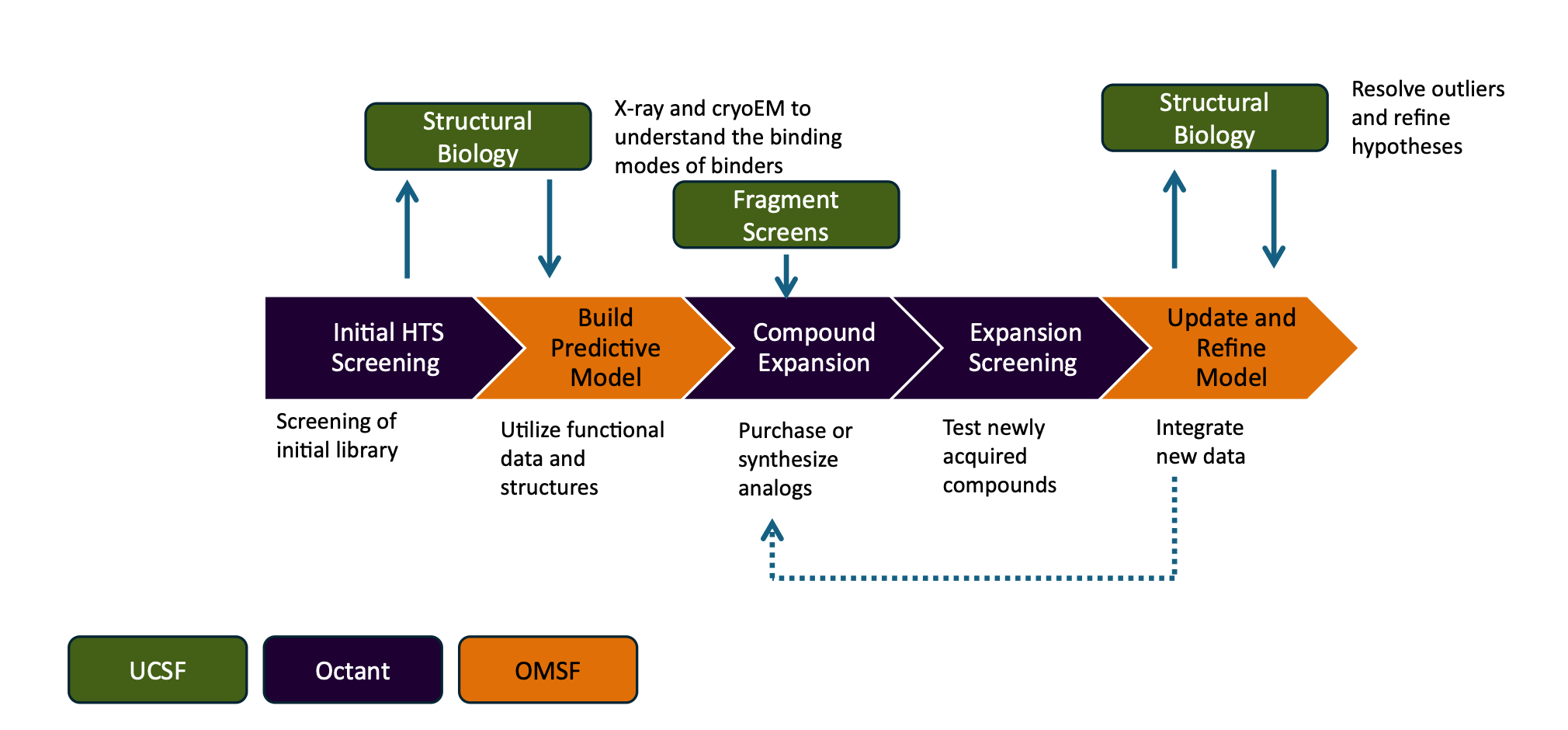
Fraser (UCSF), Edgar (Octant), Chodera (MSKCC), and Walters (OMSF) have launched OpenADMET, an ARPA-H and Gates Foundation-funded consortium generating systematic, internally consistent ADMET datasets and releasing everything publicly.
🧬 The consortium runs assays across the full “Avoid-ome” panel at industrial scale: 30,000 compounds per run in 1536-well plates, under $0.40 per compound. Active learning selects informative compounds for expansion, and structural biology (100+ PXR crystal structures so far) resolves binding modes.
⚡ This is a programme-level perspective paper rather than a single dataset release. The first community challenge has run. They’re screening tens of thousands of compounds weekly. I appreciate the honesty that this is long-term infrastructure. They’re not claiming to have solved ADMET prediction; they’re arguing nobody will until the data problem is addressed.
🧪 Where This Fits
This is an upstream data-generation effort, not a prediction tool. It sits before everything else in the ADMET pipeline: the datasets it produces will train the next generation of models. TDC (Therapeutics Data Commons) and ChEMBL aggregate existing literature data. OpenADMET’s bet is that literature data is too inconsistent to train reliable models, and that generating internally consistent measurements from scratch, with structural validation, justifies the cost. Tools like ADMET-AI (Swanson et al., 2024) would be downstream consumers of these datasets. The federated learning alternative (training behind pharma company firewalls) is explicitly discussed and dismissed: it can’t generalise beyond local chemical space and doesn’t produce the structural understanding needed for true mechanistic models.
💡 Why This Is Cool
The framing is what matters here. By defining ADMET as a finite structural biology problem (map the interactions with roughly 100 proteins and you’ve covered most failure modes), they turn an open-ended prediction challenge into a bounded experimental campaign. The question is whether $0.40-per-compound assays and active learning can produce models that generalise to novel chemical matter outside the training distribution. The open-science commitment, with Gates Foundation backing, makes this more credible than most “we’ll share data eventually” promises from pharma-adjacent initiatives.
📄 Read the paper
🗓️ Events & Competitions
The best competitions, hackathons, and community challenges in AI x life sciences, curated weekly. Know something worth featuring? Reply and let us know.
More upcoming events:
Creative Disruption Forum: Modern Drug Discovery | June 18, NIAB Cambridge
A full-day forum for biotech and R&D leaders exploring how technology is changing small molecule drug discovery. Keynote interviews with industry thought leaders followed by workshops under Chatham House Rules, limited to 60 attendees. Part of Cambridge Wide Open Week. Organised by Graham Combe and Prof Tony Sedgwick. £60 for biotech companies.
London Protein Design Day | June 23, Imperial College London
The first edition of a one-day symposium bringing together London’s protein design community and beyond. Programme spans AI-driven design, molecular dynamics, and bioinformatics, with applications across enzymes, antibodies, and materials. Organised by Pietro Sormanni, Rebecca Birolo, and Jakub Lála. Abstract deadline for poster/oral presentations is this Saturday (May 17). In person only.
BioHackathon Europe 2026 | November 9-13, Barcelona
ELIXIR’s annual international bioinformatics hackathon, running since 2018. 160+ participants, five days of collaborative coding on open bioinformatics infrastructure and tools. The call for project proposals opens March 16 and closes April 15 - so if you want to lead a project, that’s your window.
Thanks for reading!
💬 Get involved
We’re always looking to grow our community. If you’d like to get involved, contribute ideas or share something you’re building, fill out this form or reach out to me directly.
Connect With Us
Have questions or suggestions? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website
Thanks for the tools I'm going to explore and try to incorporate it...