
CNRS Lyon’s FoldScript, Tsinghau's DrugCLIP, and YCU's ChemTSv3
Kiin Bio's Weekly Insights

Welcome back to your weekly dose of AI news for life science!
What’s your biggest time sink in the drug discovery process?
🧬 FoldScript: Analysing AI-Generated Protein Structures
What if we could easily tell which AI-generated protein structures are accurate and which are hallucinated?
Artificial Intelligence is transforming the field of protein research in an unprecedented way. From predicting complex 3D structures to designing novel proteins, AI-driven research provides researchers with immediate access to highly accurate protein models. However, AI-based generative models can also invent plausible-looking structures in regions that should be unstructured, often referred to as hallucinations.
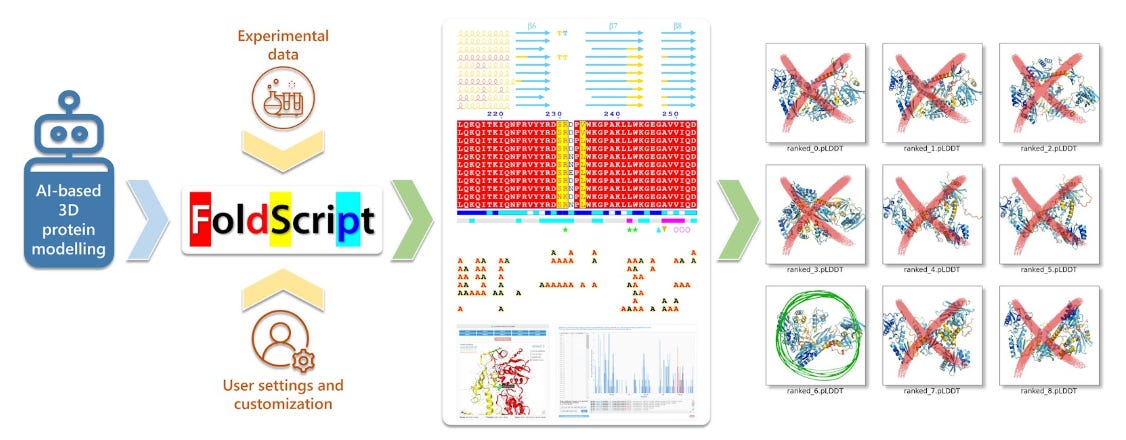
To tackle this problem, researchers based at CNRS Lyon have developed FoldScript, a web server that analyses AI-generated 3D protein models and enables selection of the most accurate output from an AI-driven protein modelling process. FoldScript allows visual inspection of structural differences and can be used by anyone without prior knowledge of structural biology.
🔬 Applications and Insights
1️⃣ Input data and large-scale analysis
FoldScript can assess up to 25 AI-generated models and accepts input formats in PDB and CIF. It gathers and synthesises information spanning primary to quaternary structure, relying on ESPript and ENDscript as standards for schematic flat-figure representations of protein structures.
2️⃣ Easy workflows
Once the data is provided, the server runs an automated pipeline that collects all information required for final display. The output is presented as clear flat figures with mosaics of 3D structures, enabling direct visual comparison of structural differences.
3️⃣ Comprehensive user interface
FoldScript offers an easy-to-read, comparative view designed for researchers, including those without structural biology expertise. Built-in tools leverage previously generated data to support rational model selection.
4️⃣ Heterogeneous data analysis
FoldScript analyses heterogeneous outputs from multiple programs quickly and automatically. Typical runtimes range from 30 seconds to 2 minutes.
💡 Why It’s Cool
FoldScript is an open platform available online and usable by anyone. Positioned upstream in AI-driven protein modelling workflows, it helps filter hallucinated structure before downstream analysis. Its speed and automation make it a practical safeguard as generative protein models become more widespread.
📄 Read the paper
⚙️ Explore the code
👏 Huge thanks to Gargi for her first contribution to Kiin Bio Weekly.
A great addition to the newsletter and excited to share more of your work with the community.
🧬 DrugCLIP: Proteome-Scale AI Screening for Undiscovered Therapeutic Targets
What if virtual screening could scale across the entire human proteome?
Roughly 90 per cent of disease-associated proteins still lack approved small-molecule therapies. One major reason is scale. Genome-wide virtual screening remains computationally impractical for traditional docking and many deep learning approaches.
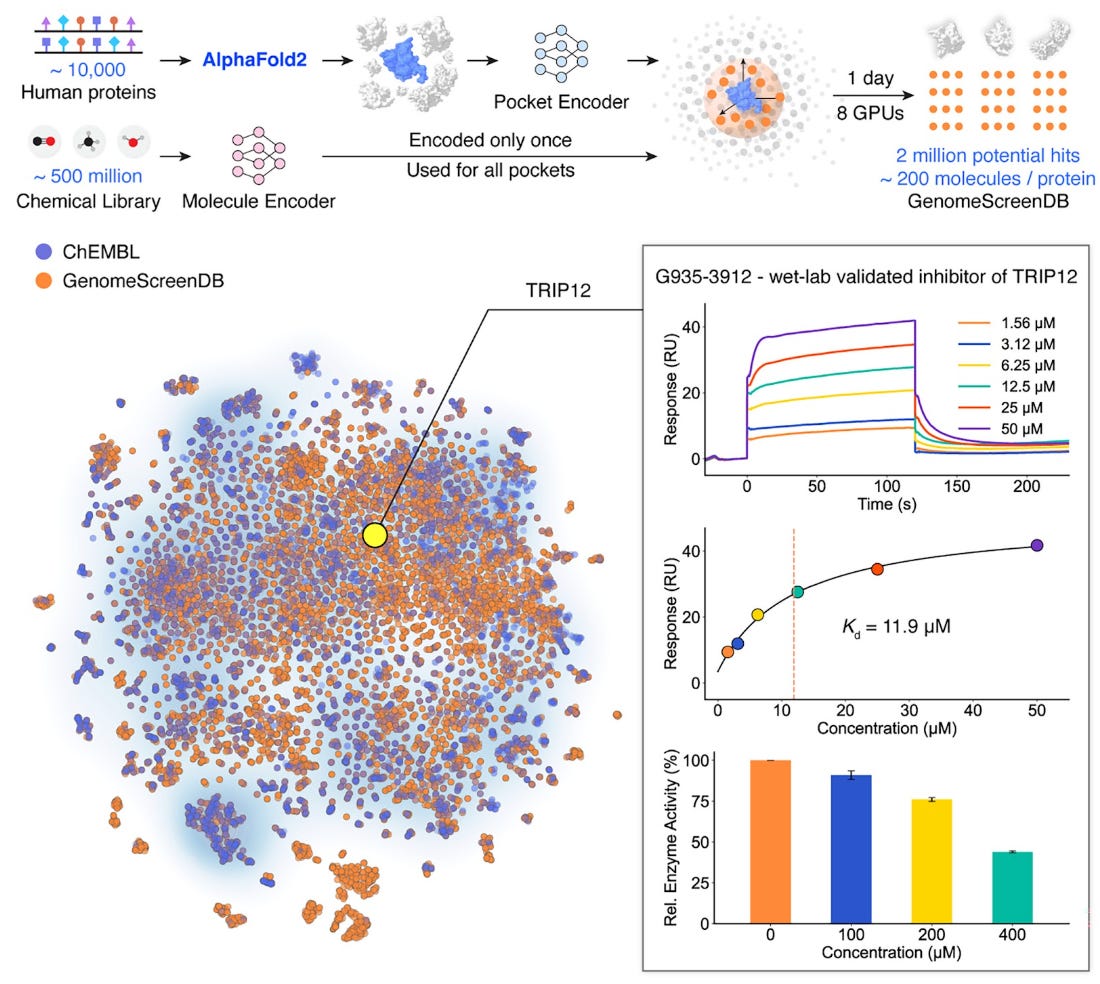
DrugCLIP addresses this limitation with a deep contrastive learning framework designed for proteome-scale screening. Instead of evaluating targets one by one, it enables simultaneous screening across the human druggable genome.
DrugCLIP embeds protein targets and small molecules into a shared latent space trained on large-scale synthetic data derived from experimentally characterised protein–ligand complexes. Crucially, it also works with AlphaFold-predicted structures through integration with GenPack, a generative refinement module that improves binding-pocket identification.
In benchmarking, around 500 million compounds were screened against approximately 10,000 human proteins in a single day.
🔬 Applications and Insights
1️⃣ Ultra-high-throughput screening at scale
DrugCLIP efficiently evaluates trillions of protein–compound pairings, turning proteome-scale screening into a baseline capability rather than a bottleneck.
2️⃣ Discovery of novel binders
Predictions were experimentally validated with meaningful hit rates, including inhibitors of the serotonin 2A receptor, the norepinephrine transporter, and THRAP12, a previously understudied protein now shown to have multiple active inhibitors.
3️⃣ Genome-informed drug discovery
Combined with GenPack, DrugCLIP enables evaluation of how genetic variation alters small-molecule binding, linking population genomics directly to early-stage discovery.
4️⃣ From ranking to discovery
Rather than refining rankings within small libraries, DrugCLIP functions as a discovery engine, generating entirely new protein–compound associations.
💡 Why It’s Cool
DrugCLIP brings the full human proteome within reach of virtual screening. It reframes AI in drug discovery from ranking molecules to uncovering targets that were previously inaccessible.
📄 Read the paper
⚙️ Explore the code
👏 Big thanks to Amber for another excellent contribution to the newsletter.
Always a great eye for interesting work and really appreciate you sharing it with our readers.
🧪 ChemTSv3: Redesigning the Search Space for Molecular Design
What if molecular design did not assume the search space was fixed?
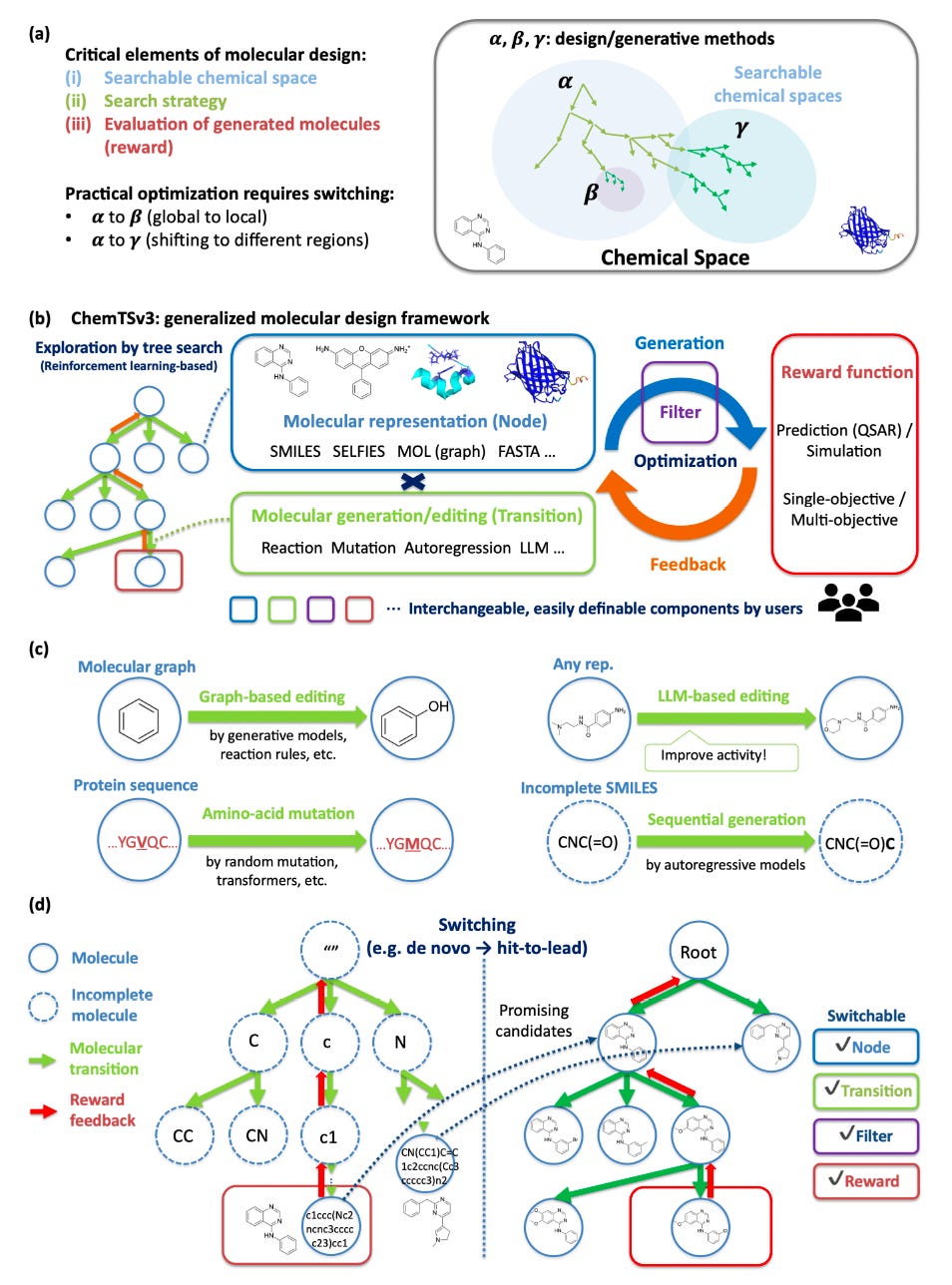
Generative molecular design often follows a familiar pattern. Broad exploration gives way to narrow optimisation, only for the original model choice to become a limitation later in the workflow.
ChemTSv3, developed by researchers at Yokohama City University, RIKEN, MolNavi, Kyushu University and the University of Tokyo, is built to resolve this tension. It treats molecular design as a search problem that can change shape mid-process.
The same framework supports different molecular representations, transition rules and optimisation strategies without restarting from scratch.
🔬 Applications and Insights
1️⃣ Dynamic search space switching
ChemTSv3 allows seamless transitions from de novo generation to local lead optimisation by swapping representations and transition rules during a single workflow.
2️⃣ One framework, many molecular forms
Small molecules, proteins and incomplete structures are treated as nodes within the same search tree. SMILES, SELFIES, molecular graphs, FASTA sequences and LLM-edited molecules coexist under one logic.
3️⃣ LLMs as editable transitions
Language models act as explicit transitions within the search rather than opaque generators. Prompts become controllable edits alongside classical graph and sequence operations.
4️⃣ Competitive performance with flexibility
On the PMO benchmark, ChemTSv3 matched or exceeded strong baselines. Dynamic search-space switching improved scores by more than two points compared with static setups.
💡 Why It’s Cool
ChemTSv3 reframes molecular generation as designing the search itself rather than tuning a single generator. It mirrors how chemists actually work: explore broadly, learn quickly, zoom in, switch tools and repeat.
📄 Read the paper
⚙️ Explore the code
Thanks for reading Kiin Bio Weekly!
💬 Get involved
We’re always looking to grow our community. If you’d like to get involved, contribute ideas or share something you’re building, fill out this form or reach out to me directly.
Connect With Us
Have any questions or suggestions for a post? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website
Foldscript might come handy in the next adaptyv bio binder design competition :p