
UPenn's mRNAutilus, GSU's EpiFormer, and Seoul National's Folddisco
Kiin Bio's Weekly Insights

Welcome back to your weekly dose of AI news for Life Science! This weeks fix:
mRNAutilus generates entire therapeutic mRNA sequences from scratch, and the wet-lab numbers are hard to argue with: 400x over wild-type expression, beating commercial Spike constructs.
EpiFormer brings geometric deep learning to epitope prediction with a 40% F1 boost. A nice complement to last week’s ESM binder design coverage, now from the antigen side.
Folddisco indexes 53 million protein structures and finds structural motifs in seconds. The Steinegger lab keeps quietly building infrastructure that makes everyone else’s work faster.
Kiin Pioneer Programme
We built a platform that helps researchers speed up their entire science, from literature review and biomarker discovery to bioinformatics and computational chemistry. If your workflow involves pulling findings from five different places before you can actually act on any of them, this is for that.
The Pioneer Programme gives academic labs and non-profits one year of free access, plus support from our science team. No cost, no data transfer, all IP stays with your institution. Applications close August, cohort starts September.

mRNAutilus: Multi-Objective-Guided Discrete Generation of mRNA with Optimized Therapeutic Properties
🔬 Designing full-length therapeutic mRNAs means optimising stability, translation efficiency, and codon usage simultaneously. Current methods tackle these objectives piecemeal, stitching together separately optimised UTRs and coding regions, which leaves performance on the table.
Patel et al. from the Chatterjee lab at Duke present mRNAutilus, a generative framework that designs complete mRNA transcripts optimised across multiple properties at once.
🧬 The system trains a masked discrete diffusion model on millions of full-length mRNAs, then steers generation with Monte Carlo tree guidance to hit multiple objectives without retraining. It operates on whole transcripts rather than modular components.
⚡ Zero-shot mRNAutilus designs encoding firefly luciferase achieved over 400-fold higher expression than wild-type, outperforming commercial baselines. For SARS-CoV-2 Spike, designs matched or surpassed both clinically used constructs and lab-optimised sequences. The framework also generalised to prime editing guides and targeted protein degradation, which suggests this is not a one-trick benchmark result.
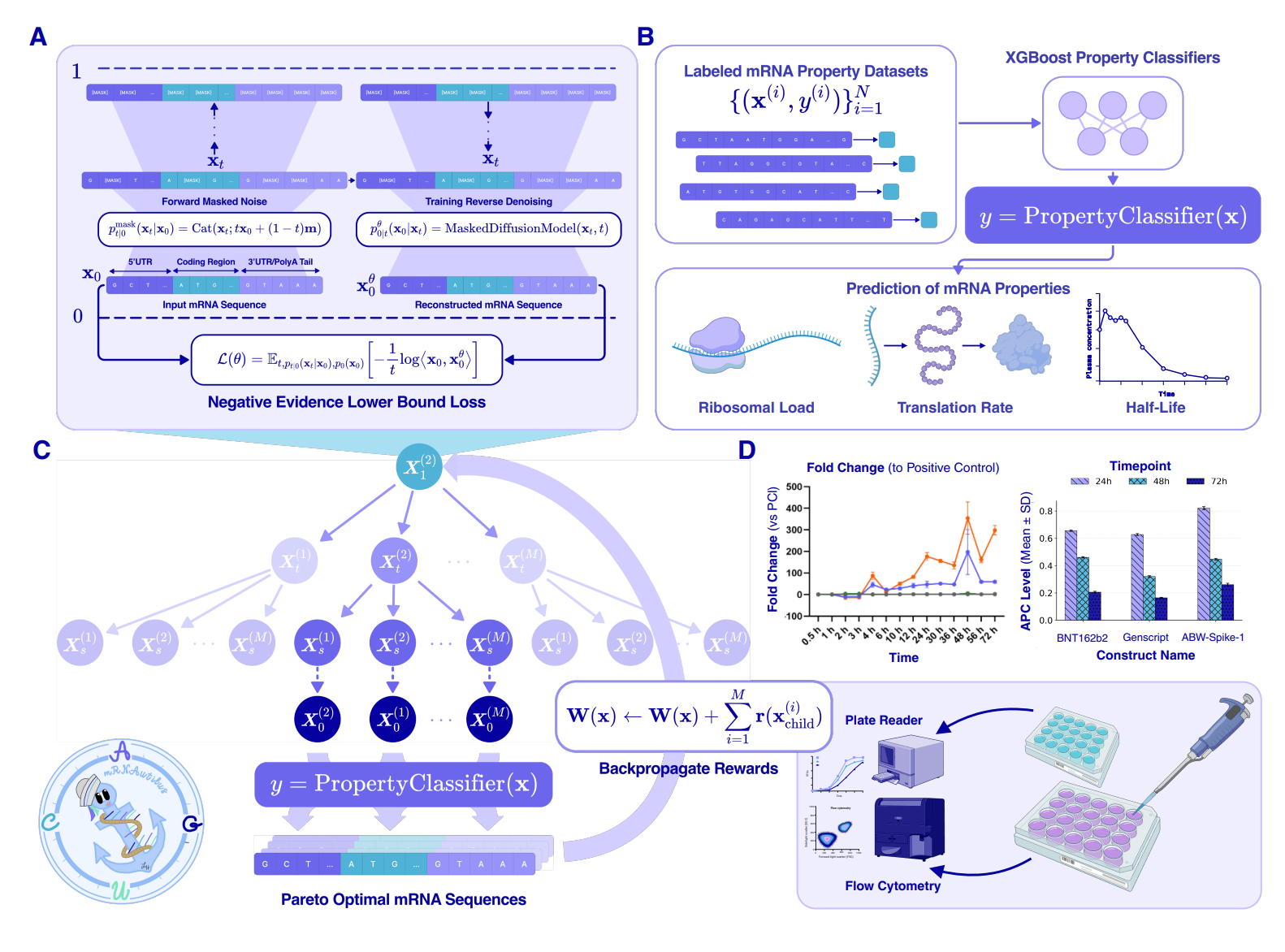
🧪 Where This Fits
mRNA therapeutics have had a sequencing problem disguised as a design problem. Post-COVID, the bottleneck is no longer “can we make mRNA drugs?” but “can we make them well enough to compete on potency and manufacturing cost?” Tools like LinearDesign (Zhang lab, 2023) optimise coding sequences for stability, and UTR-focused approaches pick regulatory elements, but these treat the transcript as a set of independent modules. mRNAutilus is the first framework I have seen that treats the entire transcript as a single generative object, which matters because interactions between UTRs and coding regions affect folding and translation in ways modular approaches miss.
The wet-lab validation is what separates this from yet another generative model paper. Beating commercial constructs for Spike expression is a meaningful bar, not an in silico benchmark. The generalisation to prime editing and degraders suggests the architecture is flexible enough to not be overfit to reporter assays. The timing makes sense too: masked diffusion models have matured enough (thanks to protein and genomics applications) that applying them to mRNA sequences is a natural next step, and the Monte Carlo tree guidance borrows from AlphaGo-era decision strategies to handle multi-objective trade-offs without expensive retraining.
For readers working in mRNA therapeutics: this is worth watching closely. The code is not yet public, which limits immediate adoption, but the approach could compress the design-test cycle considerably once available.
💡 Why This Is Cool
The shift from “optimise one property” to “generate the whole thing optimised” matters more than it sounds. The history of biologics design is littered with tools that optimised one metric while inadvertently breaking another. If multi-objective generation holds up across more constructs and delivery contexts, it moves mRNA design closer to what protein design achieved with diffusion models over the past two years. The open question is whether the approach scales to longer, more complex transcripts and novel target classes beyond the well-studied ones shown here.
📃 Read the paper.
EpiFormer: Learning Antigen-Antibody Interactions for Epitope Prediction via Geometric Deep Learning
🔬 Predicting which surface residues an antibody will target on an antigen remains stubbornly difficult. Most existing methods treat the antigen in isolation, ignoring the antibody entirely or bolting antibody information on as a late-stage afterthought.
Ahmed et al. from Georgia State University introduce EpiFormer, a geometric deep learning framework that models antigen-antibody interactions through interleaved cross-attention within GNN encoding layers.
🧬 Rather than encoding antigen and antibody separately then combining representations at the end, EpiFormer threads cross-attention between the two structures at every encoding layer. This allows bidirectional information flow throughout the representation, so the model learns how antibody geometry constrains which epitope residues are accessible.
⚡ On standard benchmarks, EpiFormer achieves over 40% improvement in F1 score compared to previous best methods. That is a substantial jump for a prediction task where incremental gains of 2-5% have been typical. The model operates on 3D structural inputs from antibody-antigen complexes.
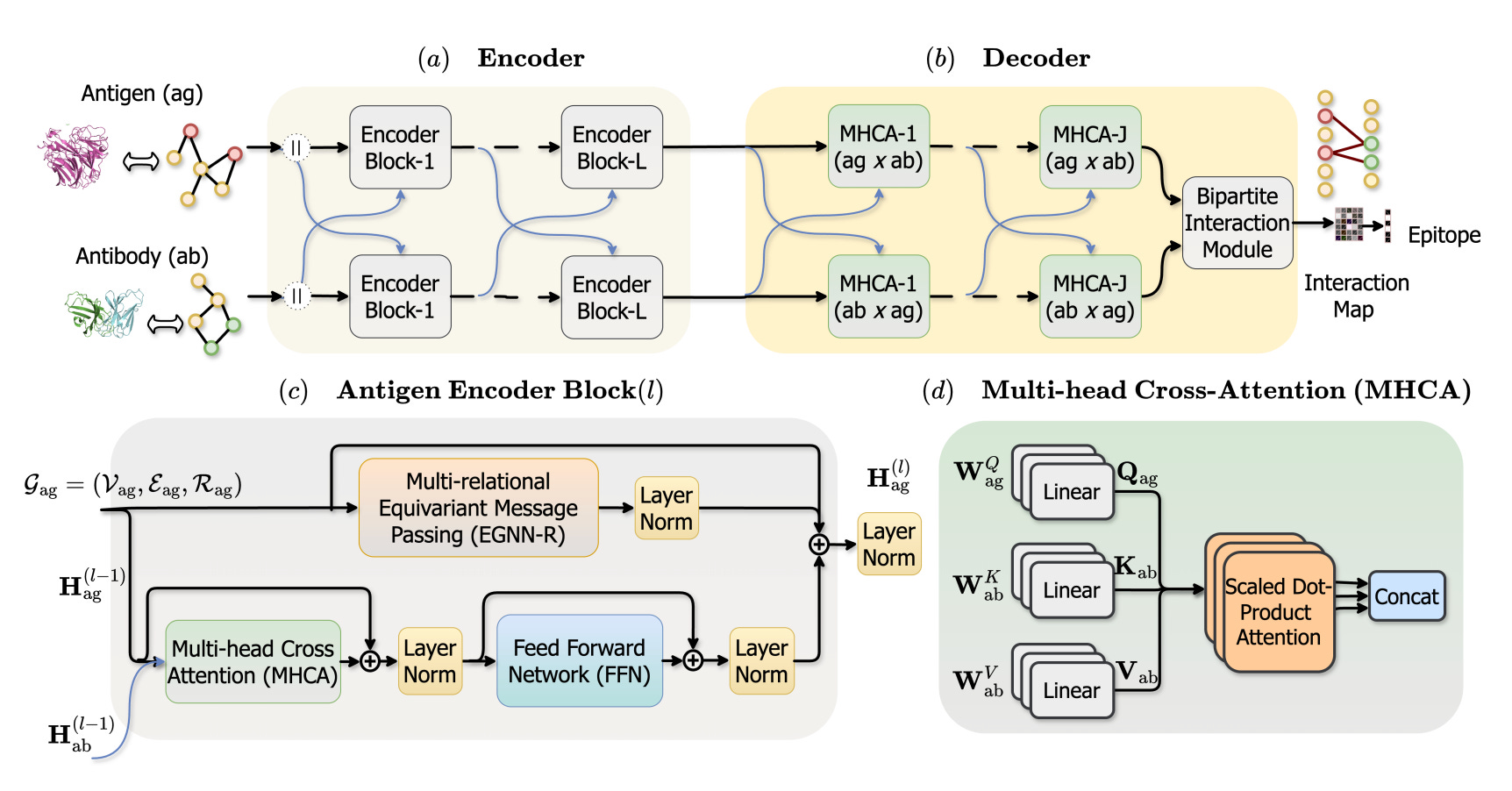
🧪 Where This Fits
Epitope prediction sits upstream of antibody engineering: if you know where an antibody binds, you can design better binders and prioritise vaccine targets. Previous approaches like DiscoTope and ElliPro use geometry and surface properties of the antigen alone, which is a bit like predicting where a key fits without looking at the lock. More recent methods (PECAN, AsEP) incorporated paratope information but typically as a separate encoding step with late fusion.
EpiFormer’s contribution is architectural rather than data-driven. The interleaved cross-attention ensures antibody context informs antigen representations from the start rather than being concatenated at the decision layer. This connects naturally to last week’s coverage of ESM-based binder design: that work generates antibodies given a target, while EpiFormer predicts where on the target those antibodies will land. Together they cover both directions of the same binding prediction problem.
The 40% F1 improvement is striking, though it warrants some caution. Epitope prediction benchmarks are notoriously sensitive to train/test splitting, and structural epitope datasets remain small (a few thousand complexes in SAbDab). Whether this holds on truly novel antigen folds or just reflects better exploitation of known structural patterns is an open question. The code is available, which helps.
💡 Why This Is Cool
The “interleave information early rather than fuse late” lesson keeps appearing across structural biology. AlphaFold did it for MSA and structure tracks. ESM3 does it for sequence, structure, and function. EpiFormer applies the same intuition to a paired prediction problem. The field has been underestimating how much cross-modal information gets lost in late-fusion architectures, and each new result in this direction makes that clearer. For antibody discovery teams, this is immediately useful if it generalises beyond the benchmark setting.
📃 Read the paper.
💻 Try the code.
Structural Motif Search Across the Protein Universe with Folddisco
🔬 Finding recurring 3D structural motifs (zinc fingers, catalytic triads, protein-protein interaction surfaces) across millions of predicted structures is computationally prohibitive. Existing methods either cannot handle discontinuous motifs or choke on databases beyond a few hundred thousand structures.
Kim et al. from the Steinegger lab at Seoul National University present Folddisco, a structural motif search tool that indexes 53 million AFDB50 structures in a 1.45 TB index and returns query results in seconds.
🧬 Folddisco encodes proximal residue pairs into geometric feature sets (distances, angles, and side-chain orientation via torsion angles), stores them in a position-independent inverted index, and ranks hits using an IDF-based coverage score that rewards rare features. This handles both short continuous motifs and long discontinuous ones.
⚡ Indexing is 11x faster to build and 4x more storage-efficient than previous state-of-the-art. Query speed is 20-fold faster than pyScoMotif on the full pipeline. On the zinc finger benchmark against the human proteome, Folddisco outperformed both RCSB and pyScoMotif on recall while maintaining higher precision. It also successfully distinguished active from inactive GPCR conformational states using activation motifs.
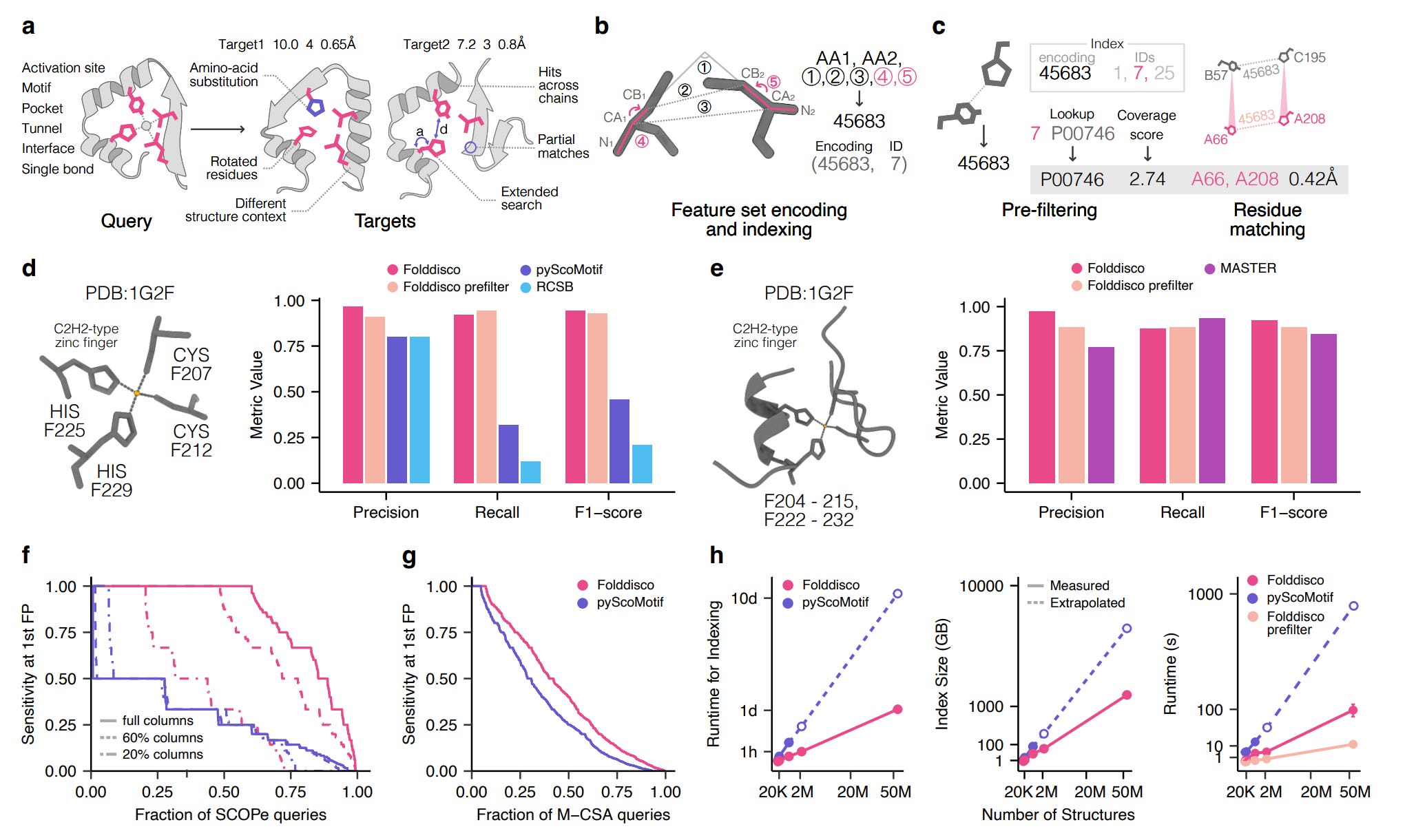
🧪 Where This Fits
This is infrastructure work, and the most consequential kind. The AlphaFold database gave us 200+ million predicted structures, but searching them structurally has lagged far behind searching them by sequence (where tools like Foldseek, also from the Steinegger lab, already operate at scale). Folddisco fills the motif-search gap: given a 3D pattern of interest, find everywhere it occurs across all known and predicted protein structures.
The practical value becomes clear with the GPCR example. Being able to query “show me all structures with this activation motif” across both experimental PDB structures and AlphaFold predictions means you can study conformational states at proteome scale. That was previously manual curation work. Similarly, the zinc finger detection in uncharacterised metagenomic proteins (from ESM30) demonstrates functional annotation where sequence-based methods fail entirely.
Folddisco’s main limitation is its 20-angstrom connectivity constraint, which means it cannot detect spatially distant functional sites like remote allosteric pockets. The IDF scoring also struggles with very short motifs. These are known trade-offs for the speed gains.
💡 Why This Is Cool
The Steinegger lab has been building the search infrastructure for the structure-prediction era piece by piece: MMseqs2 for sequences, Foldseek for structure alignment, and now Folddisco for motif search. Each tool makes the previous one more useful. What matters here is not the individual benchmarks but the fact that motif search at 53-million-structure scale is now a webserver query rather than a compute cluster job. That means anyone with a structural intuition and a browser can generate hypotheses that previously required a compute cluster and custom code. The webserver is live at search.foldseek.com/folddisco.
📃 Read the paper. 💻 Try the code.
🗓️ Events & Competitions
The best competitions, hackathons, and community challenges in AI x life sciences, curated weekly. Know something worth featuring? Reply and let us know.
More upcoming events:
Creative Disruption Forum: Modern Drug Discovery | June 18, NIAB Cambridge
A full-day forum for biotech and R&D leaders exploring how technology is changing small molecule drug discovery. Keynote interviews with industry thought leaders followed by workshops under Chatham House Rules, limited to 60 attendees. Part of Cambridge Wide Open Week. Organised by Graham Combe and Prof Tony Sedgwick. £60 for biotech companies.
London Protein Design Day | June 23, Imperial College London
The first edition of a one-day symposium bringing together London’s protein design community and beyond. Programme spans AI-driven design, molecular dynamics, and bioinformatics, with applications across enzymes, antibodies, and materials. Organised by Pietro Sormanni, Rebecca Birolo, and Jakub Lála. Abstract deadline for poster/oral presentations is this Saturday (May 17). In person only.
BioHackathon Europe 2026 | November 9-13, Barcelona
ELIXIR’s annual international bioinformatics hackathon, running since 2018. 160+ participants, five days of collaborative coding on open bioinformatics infrastructure and tools. The call for project proposals opens March 16 and closes April 15 - so if you want to lead a project, that’s your window.
Thanks for reading!
💬 Get involved
We’re always looking to grow our community. If you’d like to get involved, contribute ideas or share something you’re building, fill out this form or reach out to me directly.
Connect With Us
Have questions or suggestions? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website