
Dyna-1: Learning protein dynamics from what's missing
Deep Dive | Edition 5


Welcome back to the deep dive, where we break down the AI tools and data reshaping how new drugs are discovered. In each edition, we speak directly with the teams behind these tools to explain what they solve, how they work and where they are going next.
Today we are taking a look at Dyna-1, a deep learning model developed jointly by Gina El Nesr at Stanford University and Hannah Wayment-Steele at University of Wisconsin-Madison. We spoke with Gina, to hear the story behind the tool. Gina is currently a PhD candidate in Biophysics at Stanford; she previously received her bachelors degrees in computer science, biophysics, and applied math and statistics from Johns Hopkins University. Gina is also currently the chair organiser of the Machine Learning for Structural Biology Workshop at NeurIPS, a cornerstone conference for the community.
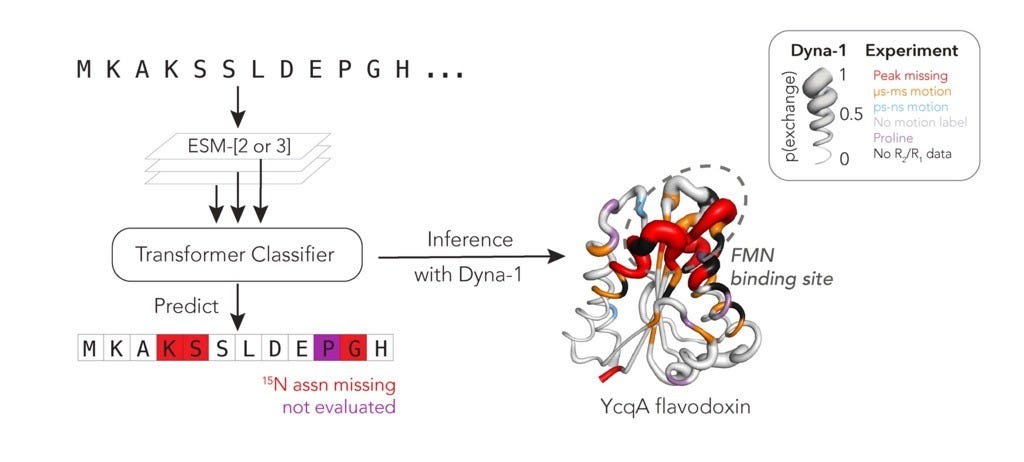
That blend of computation and structural biology sits at the heart of this project. Dyna-1 learns something remarkable about proteins not from what is present in NMR data, but from what is missing.
🔴 The Problem
Proteins are not static. Their function often depends on movements that happen on the microsecond to millisecond (µs-ms) timescale, which includes motions underlying enzyme catalysis, ligand binding, and allosteric signaling. This is exactly the timescale that has been hardest to study computationally.
As Gina explained to us, the field lacks for protein dynamics the equivalent of what the Protein Data Bank provided for AlphaFold-2: big data. Most molecular dynamics simulations capture only femtosecond to hundreds of nanoseconds of dynamics, typically too fast to observe biologically important events. Nuclear magnetic resonance (NMR) reaches the right timescales, but Biological Magnetic Resonance Data Bank (BMRB) deposits are small and inconsistent across experiments, conditions, and formats, so they are hard to use as a unified training signal. Without standardised, large-scale data on dynamics, deep learning had no clear foundation for protein dynamics at this timescale.
💡 The Idea
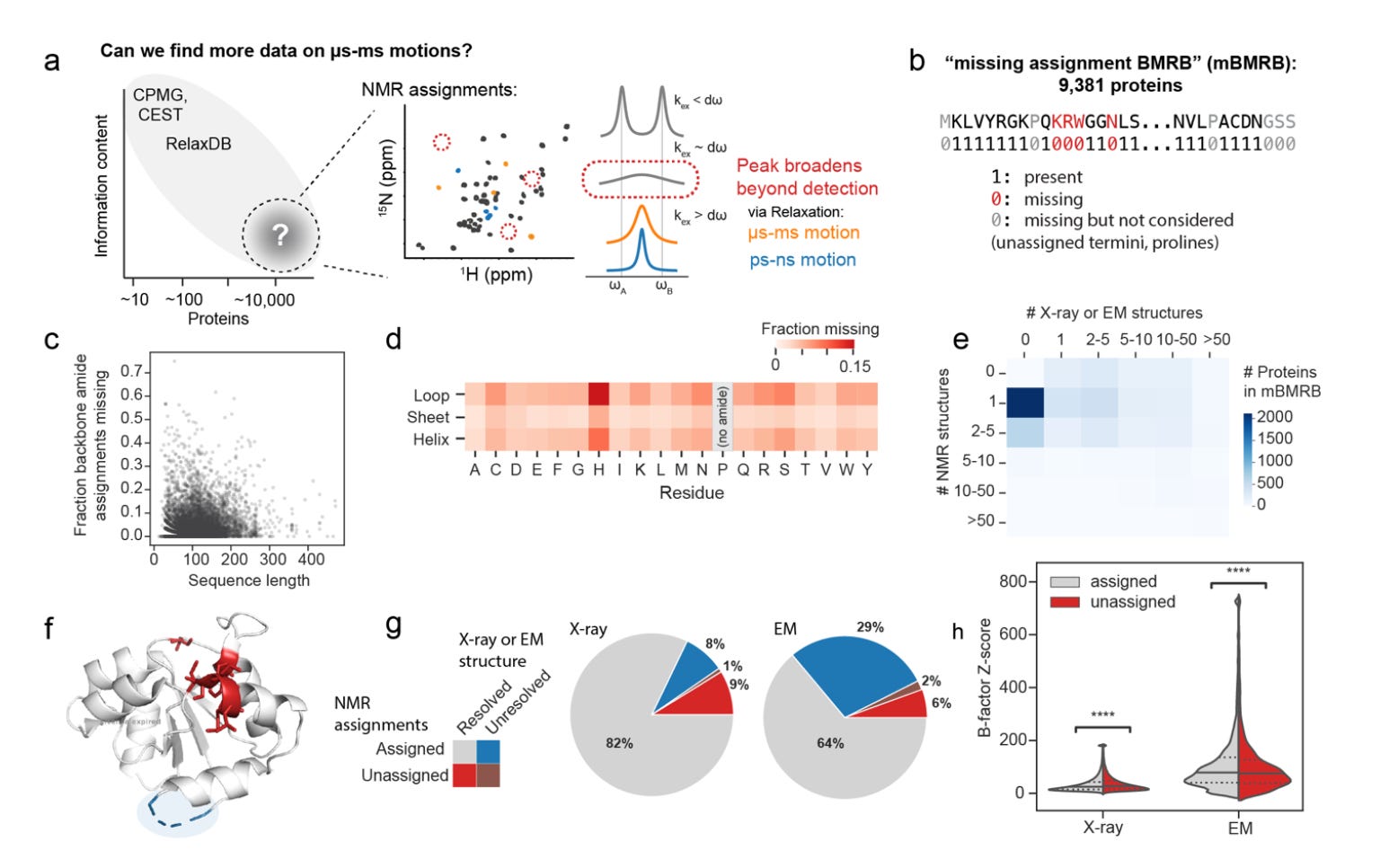
The breakthrough came from looking at what is missing in NMR spectra. Some residues that cannot be assigned might be missing for a number of reasons; however, one systematic reason a peak might disappear and not be assigned is because the residue is exchange-broadened, flipping between states on the µs-ms timescale.
Before landing on that signal, they spent more than a year trying to assemble reliable labels for slow dynamics from the BMRB and literature, even digitising points from published plots because raw datasets were rarely deposited. I can’t even begin to imagine what that process must have been like. Brutal. That effort underscores how little standardised training data existed. Their dataset became RelaxDB, a benchmark dataset on dynamics consisting of 133 proteins.
While analyzing RelaxDB, they realized that there is actually another label in the datasets they had; the residues missing assignments! They then turned to the BMRB and treated missing amide assignments as proxy labels for slow exchange. By carefully curating all the deposited proteins and filtering out confounders such as isotope labelling, they built the largest dynamics dataset for this slow timescale to date, the mBMRB, at roughly 9,400 proteins.
On top of this, they trained a classifier using embeddings from ESM-3, a multimodal protein language model. Their resulting model, Dyna-1, was trained on the missing assignment label, but generalizes beyond this to predict whether a residue undergoes slow dynamics often at biologically relevant sites. They even demonstrated it has predictive power on Rex from CPMG NMR experiments.
📊What about the training data?
Dyna-1 learns from absence rather than presence. Training and evaluation draw on two sources that the authors also curated for this work:
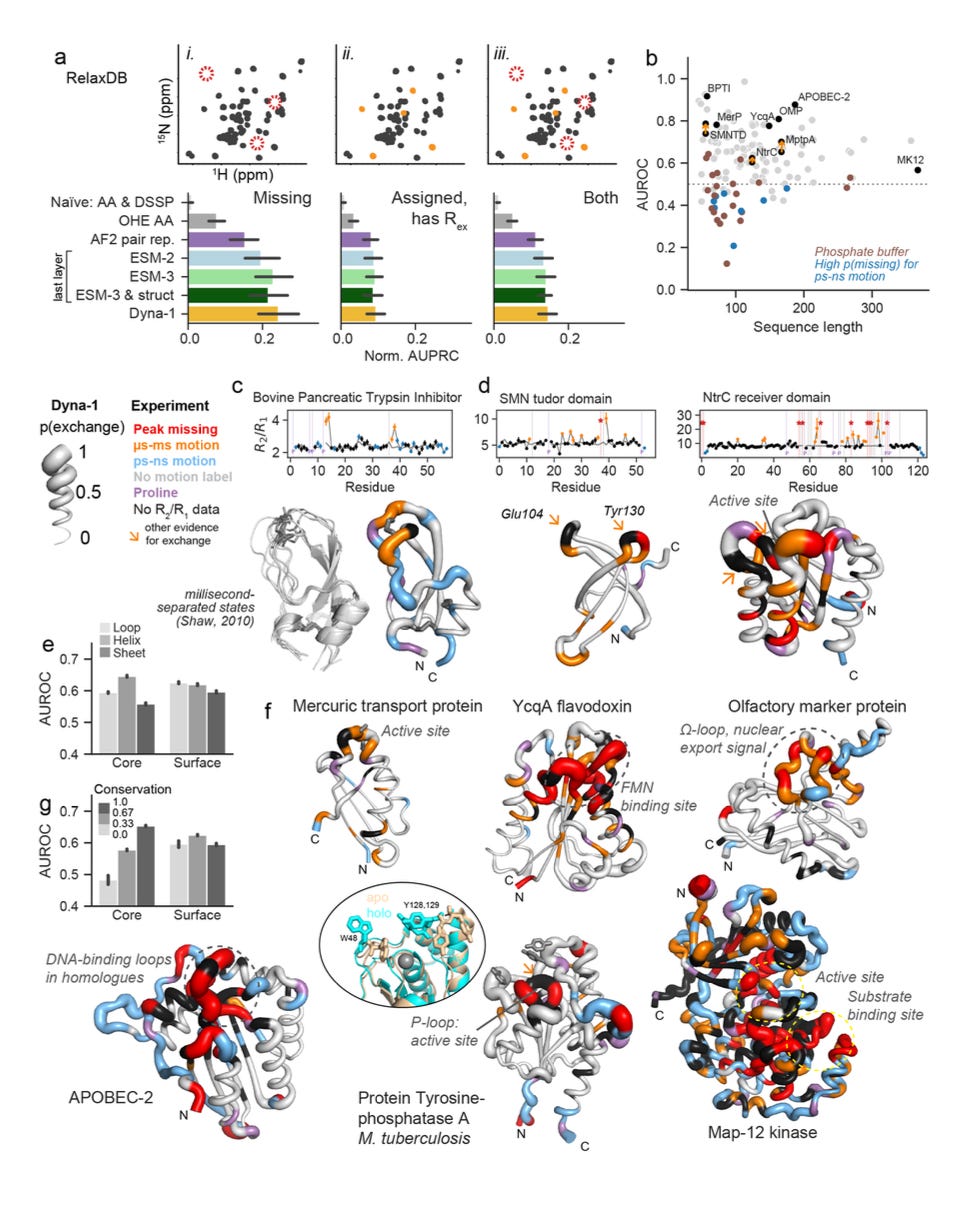
RelaxDB: 133 proteins with standardised relaxation labels, used to benchmark predictions against residue-level Rex and related readouts.
mBMRB: 9,381 proteins labelled by presence or absence of 15N backbone assignments after stringent curation to reduce confounders. These labels scale two orders of magnitude beyond RelaxDB and were used to train Dyna-1
Their curated datasets are public, RelaxDB have been released openly so that others can build on the work. The mBMRB will be made public soon.
🔬 Why It’s Different
Learning from absence: Most models learn from positive data. Dyna-1 instead extracts signals from missing data across thousands of depositions.
Validation across methods: Predictions matched not only missing assignments but also Rex values from relaxation data and CPMG experiments. In some cases, Dyna-1 flagged dynamics that conventional analysis had overlooked, which were later validated by looking back in the raw data.
Functional hotspots: The model’s high-confidence predictions often mapped onto catalytic loops, ligand-binding sites, or conserved allosteric regions, showing it was also finding biologically meaningful motions.
Architecture choice: The best performance came from an intermediate layer of ESM-3, which suggests that dynamics information is already best embedded in a model that encodes both protein sequence and structure.
Most significantly, Dyna-1 is currently the only model operating at this timescale!
🔮 The Future
The Dyna-1 work really opens the door to a new generation of tools that treat protein dynamics as a core constraint in how we study and engineer proteins, which undoubtedly will be very interesting to watch evolve across the coming years.
Better data will be key. It could allow us to expand beyond backbone amides to sidechains, larger proteins, and eventually raw spectra to sharpen our predictive power.
New benchmarks could emerge if more labs deposit their dynamics data in standardised form, just as AlphaFold benefited from community-wide sharing of protein structure and sequence data.
Design applications are already clear. A model like Dyna-1 could for example help protein engineers zero in on dynamic allosteric sites without running time-consuming NMR experiments.
If AlphaFold proved the power of protein structure, Dyna-1 points to the next frontier: understanding proteins as moving systems. The hope (I imagine) is that in the near future, dynamics models will be as central to biomedicine as structure models are today.
📄 Read the paper!
⚙️ Try it out on GitHub.
👾 Check it out on Colab.
👩🔬 Get in touch with Gina.
Thanks for reading!
Did you find this newsletter insightful? Share it with a colleague!
Subscribe now to stay at the forefront of AI in Life Science and keep up with this upcoming season of deep dives.
Connect With Us
Have questions or suggestions for our next deep dive? We’d love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website