
Epic's CoMet, Genentech's IGLOO, and Geneva's Bottom-Up CVs for Structure-Aware Biomedicine
Kiin Bio's Weekly Insights

Welcome back to your weekly dose of AI news for Life Science!
What’s your biggest time sink along the drug discovery process?
CoMet: Foundation-Scale Modeling for Clinical Event Forecasting
Epic trained a healthcare foundation model on 115 billion events.
CoMET (Cosmos Medical Event Transformer) is a large-scale transformer designed to model real-world clinical timelines across millions of patients. It predicts diagnoses, labs, medications, and hospital outcomes all without task-specific fine-tuning.
Built on data from 118 million patients in the Epic Cosmos dataset, CoMET treats longitudinal EHR data like a language of medical events. It learns to generate and forecast patient journeys directly, across specialties, care settings, and time.
Most importantly, it works. Despite never being fine-tuned, CoMET delivers strong performance across a wide range of clinical tasks, from hospital readmission to disease onset. It’s an exciting release for anyone building predictive tools, simulations, or clinical reasoning systems from real-world EHR data.
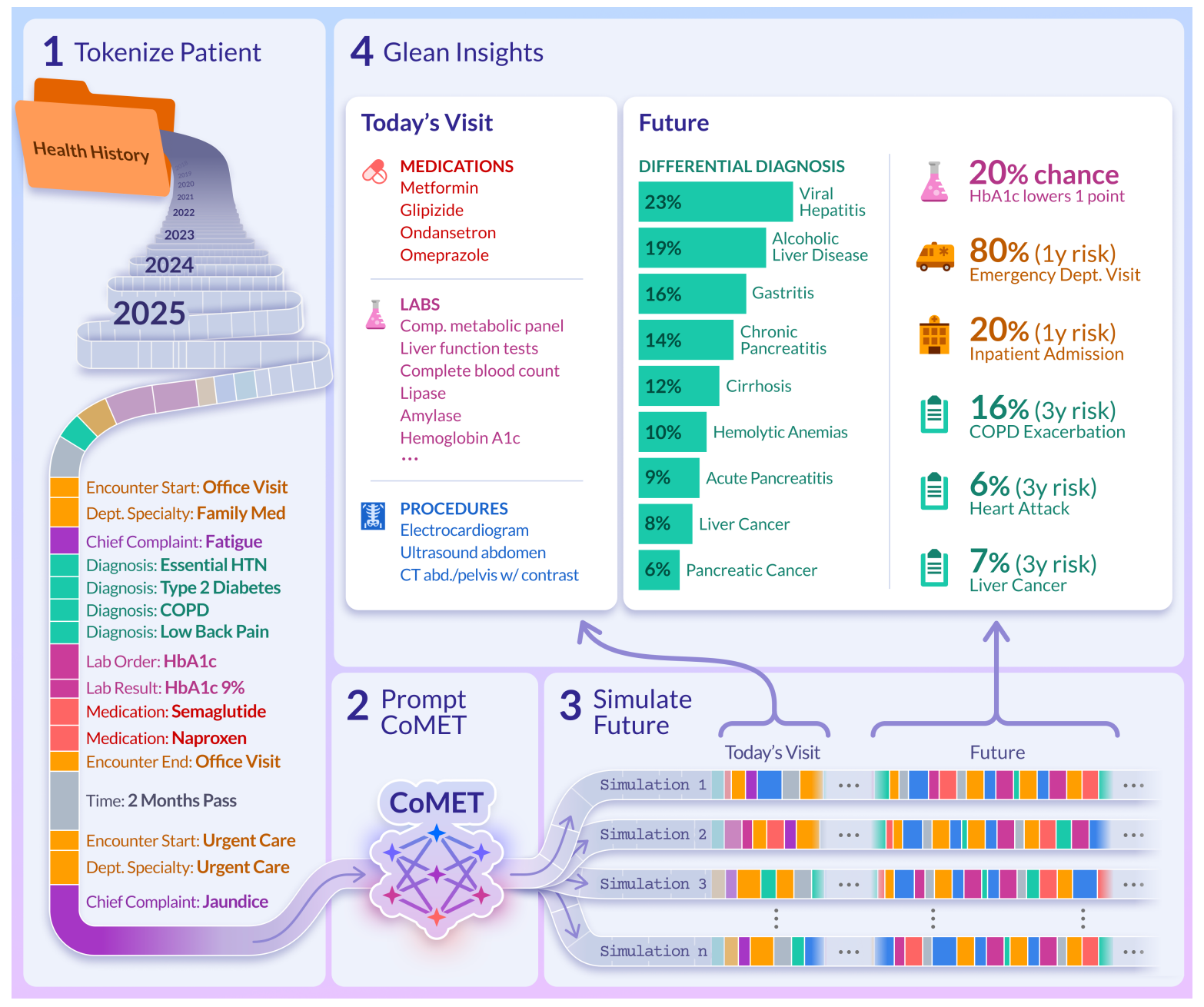
Applications and Insights
1. Unified modelling across modalities
CoMET tokenises structured EHR events, diagnoses, procedures, vitals, labs, and prescriptions into a unified sequence. It learns to generate what happens next, whether that is a new diagnosis, a lab shift, or a different encounter type.
2. Strong zero-shot performance
Across 10 clinical tasks, CoMET achieves AUROC scores between 0.77 and 0.97. This includes disease prediction (CHF, AFib, T2D), hospital outcomes (readmission, length of stay), and encounter forecasting all without fine-tuning.
3. Scales with data and parameters
Larger versions of CoMET, up to 1 billion parameters, consistently perform better. Scaling data volume also improves results, showing the value of broad patient coverage and diverse care settings.
4. Simulates realistic patient timelines
CoMET can generate full synthetic patient journeys that reflect real-world patterns like multimorbidity and care transitions. These are useful for simulation, training, and privacy-safe analysis.
I thought this was cool because it suggests a broader shift is on the horizon in clinical AI. Instead of building one-off risk models, people are beginning to train general-purpose systems that understand how care actually unfolds over time.
CoMET does not just learn a single task. It learns the statistical structure of healthcare itself. So it looks at how people get sick, how they interact with the system, and how outcomes develop. I think that’s the kind of foundation modern health systems need.
As it’s trained on 115 billion events from across the real world, it captures the complexity, messiness and scale that defines actual clinical practice.
📄Check out the paper!
Physically-Inspired CVs for Faster and Clearer Protein Folding Simulations
New collective variables make protein folding simulations faster and more detailed.
A new study from the University of Geneva introduces a practical way to build physically meaningful collective variables (CVs) that improve enhanced sampling for protein folding.
The method focuses on two features that are crucial but tend to be overlooked:
1. Hydrogen bonds, distinguishing protein–protein from protein–water interactions
2. Side-chain packing, including both native and non-native contacts
By combining these features, the authors designed CVs that help simulations move efficiently across the folding landscape, resolving not just the folded and unfolded states, but also the important intermediates in between. Pretty cool.
They tested the approach on two classic benchmark proteins, Chignolin and TRP-cage, and got accurate folding free energies, faster convergence, and much clearer resolution of subtle structural differences.
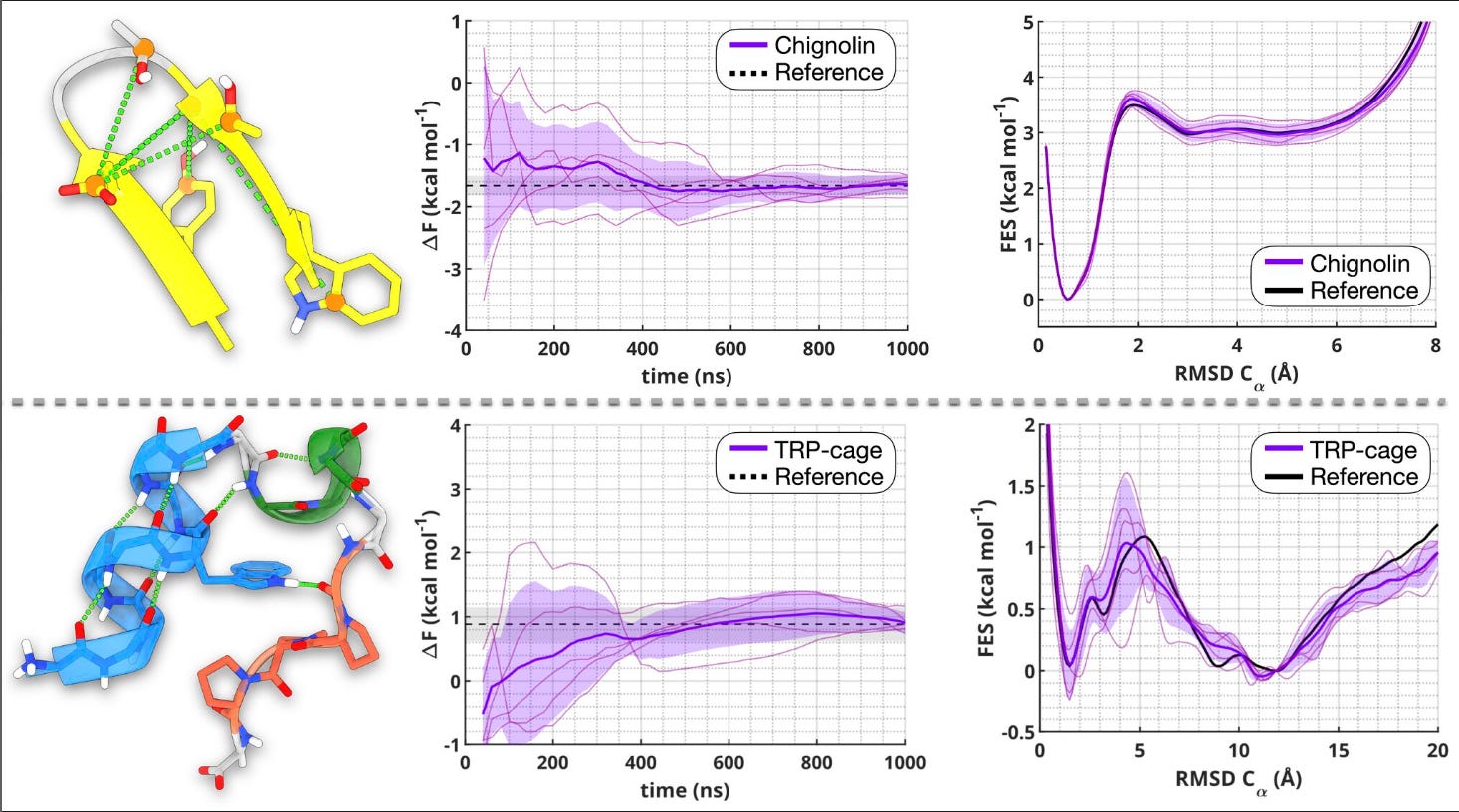
Applications and Insights
1. Captures important folding forces
The two CVs (called sHB and sSC) track hydrogen bonding and side-chain compactness in detail. They capture the interactions that really drive folding, including those involving water which is something many older CVs miss.
2. Matches long unbiased simulations
Using just short trajectories as input, the method produced free-energy estimates that matched those from simulations hundreds of microseconds long. For TRP-cage, the predicted folding energy was within 0.05 kcal/mol of the reference.
3. Reveals hidden intermediate states
These CVs can distinguish between disordered, misfolded, and partially folded states including rare intermediates like the dry molten globule, which are hard to detect with traditional metrics like RMSD.
4. Easy to use and adapt
The full workflow is automated. You just need short simulations of the folded and unfolded states, and the code outputs files ready for enhanced sampling tools like PLUMED. It’s portable, interpretable, and set up for scaling to bigger proteins.
I thought this was cool because it solves two fairly common problems in folding simulations: slow convergence and low resolution. You get speed, but you also get clarity.
Instead of just reaching the folded state, this approach shows you how you got there: through real intermediates, not just noise. And because the CVs are based on real physical features, they’re easy to understand and generalise. For anyone working on protein folding, enhanced sampling, or CV development, this is a pretty exciting solid and useful step forward.
📄Check out the paper!
⚙️Try it out the code.
IGLOO: Structure-Aware Tokenisation for Antibody Loop Design
A new tokeniser brings structure awareness to antibody loops.
A new study from Genentech introduces IGLOO, a learned representation of antibody loops that captures both sequence and structure and makes them compatible with modern protein language models. Exciting.
The method focuses on two things that matter but are hard to model:
1. The backbone geometry of flexible loops, especially those like H3 that don’t fit standard clusters
2. The sequence context, which drives subtle structural differences in binding regions
By combining contrastive learning with a dihedral-based loss, the authors trained a model that groups loops by structure, even when sequences differ. So this lets you retrieve, compare, or generate loops in a format that works with downstream models. Pretty clever.
They tested the approach on loop retrieval, binding prediction and loop generation. In every case, IGLOO outperformed existing models, despite being smaller and more targeted.
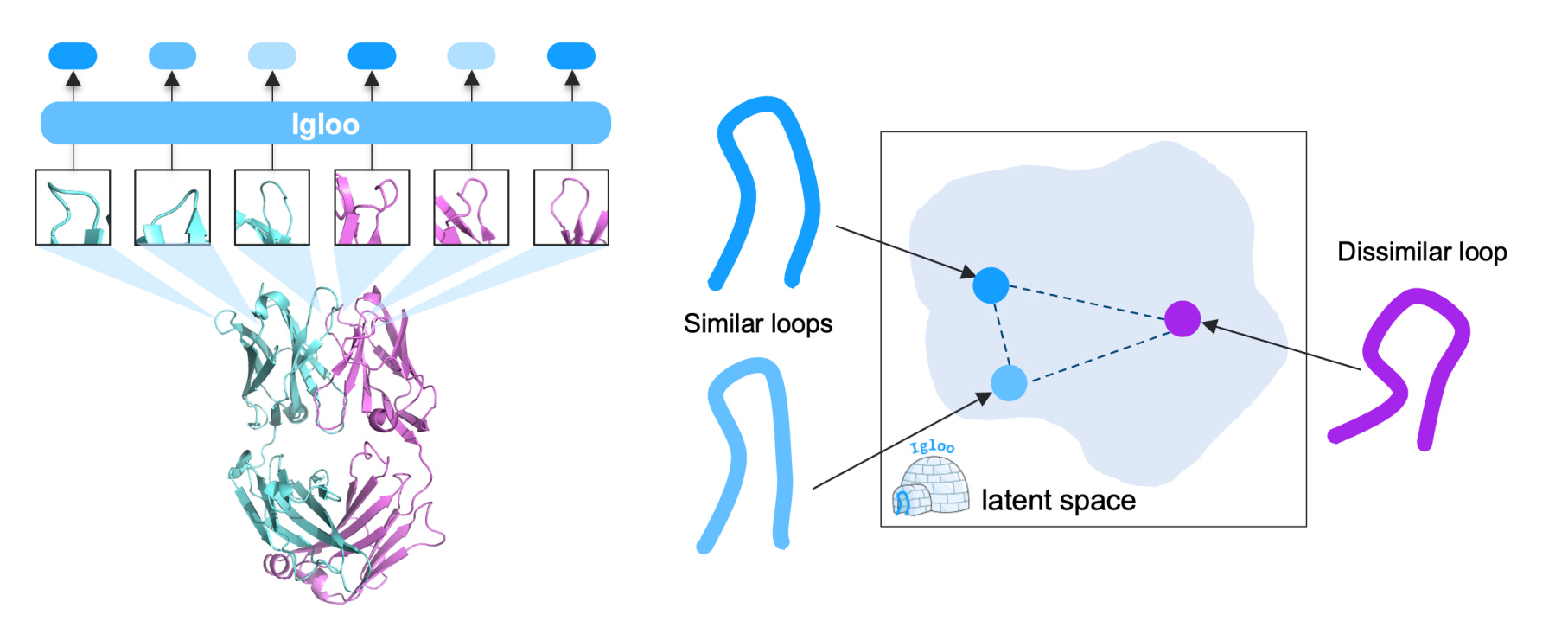
Applications and Insights
1. Learns structural loop similarity
Using a dihedral-based contrastive loss, IGLOO maps loops with similar 3D structure close together. It improves loop retrieval performance by 5.9% over the best prior model on H3 loops.
2. Boosts antibody binding prediction
Tokens from IGLOO, when used in a model called IGLOOLM, improved antibody-antigen binding prediction across 10 datasets. It beat ESM-2 in 8 out of 10 cases, despite being 7 times smaller.
3. Enables diverse yet realistic loop generation
IGLOOALM, a generative model built on IGLOO tokens, produces new loops that match target structures while exploring sequence diversity. It outperforms inverse folding baselines like AbMPNN.
4. Compatible with foundation models
As the tokens are compact and general, IGLOO can plug into antibody LMs like IgBert or be used as structure-aware inputs in design workflows without relying on hand-crafted loop classifications.
I thought this was cool because it solves one of those quiet but widely and annoyingly persistent challenges in antibody modelling. Loops are structurally diverse and hard to work with, but they are essential to function.
IGLOO makes it easier to learn from them, design with them, and scale those workflows. It gives antibodies the kind of structure-aware tokenisation that has been missing, and it fits right into the kinds of models people are already using.
For anyone working on antibody design, structure-based learning, or generative tools, this is an interesting step forward, which I’m sure many will be testing out for themselves.
📄Check out the paper!
⚙️Try it out the code.
Thanks for reading!
Did you find this newsletter insightful? Share it with a colleague!
Subscribe Now to stay at the forefront of AI in Life Science.
Connect With Us
Have questions or suggestions? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website