
ETH Zurich’s AMP-BMS/MM, CTU Prague’s OPIA, and Planck’s AlphaDIA
Kiin Bio's Weekly Insights

Welcome back to your weekly dose of AI news for Life Science!
What’s your biggest time sink in the drug discovery process?
🧪 AMP-BMS/MM: Protein-Scale Molecular Dynamics at Near-DFT Accuracy
Can we finally simulate protein-scale chemistry at DFT accuracy, in water, at scale, without breaking the bank?
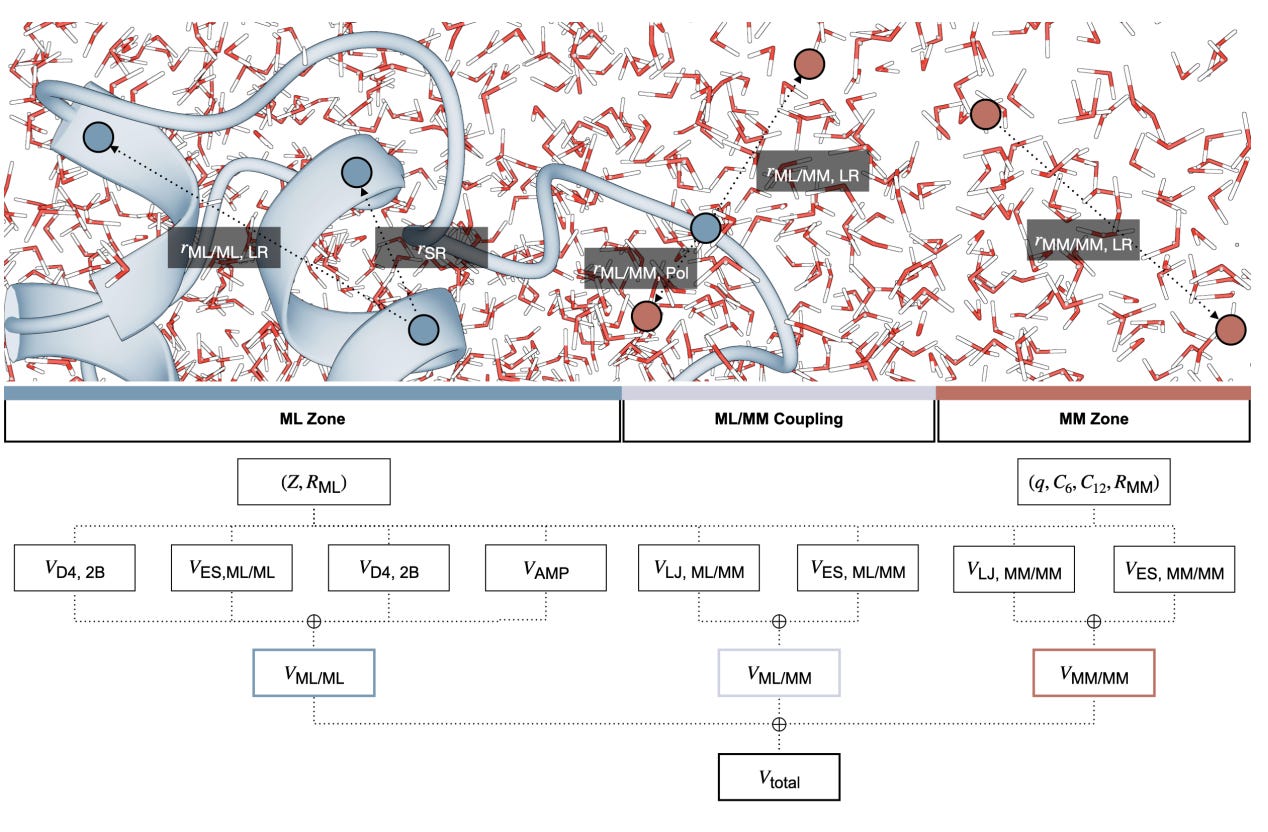
That is what AMP-BMS/MM, developed by researchers at ETH Zurich, Mila and Roche, sets out to demonstrate. It is a physics-aware ML/MM hybrid architecture that combines directional neural network potentials, explicit solvent, and enzyme-scale simulations trained on data designed for real biochemistry rather than vacuum fragments.
The headline claim is ambitious: 100 ns molecular dynamics simulations of full proteins with near-DFT accuracy, in solution, running on a single GPU.
🔬 Applications and Insights
1️⃣ Protein dynamics at scale without force fields
AMP-BMS/MM simulates full proteins in explicit water using ML accuracy rather than classical force fields. RMSDs stabilise around 1–1.5 Å over 100 ns, comparable to high-quality force fields but without parametrisation.
2️⃣ Captures intrinsically disordered regions and folding energetics
The model reproduces experimental melting curves for small proteins and disordered peptides. It avoids the common over-stabilisation seen in classical models by correctly balancing solute–solvent interactions.
3️⃣ Full-enzyme reaction barriers without truncated QM regions
Simulations of chorismate mutase and fluoroacetate dehalogenase treat the entire enzyme within the ML region. No link atoms, no reduced QM boxes. Barrier shifts between mutants closely match experiment.
4️⃣ Built for physics, optimised for speed
The architecture uses anisotropic message passing and is trained on the new BMS25 QM/MM dataset. Electrostatics, dispersion and polarisation are handled explicitly, and the model scales to hundreds of thousands of atoms.
💡 Why It’s Cool
This is not another fragment-trained neural potential. AMP-BMS/MM models biological chemistry as it actually happens, with long-range physics, explicit solvent and no hard-coded shortcuts. It is fast, general and ready to scale.
📖 Read the paper
💻 Try the code
🧬 OPIA: Learning Protein Function From a Single Sequence
What if you did not need millions of sequences to train a protein model?
Protein language models typically rely on massive datasets, alignments and evolutionary histories. But researchers at Czech Technical University in Prague, Seoul National University and MIT show that much of that information can come from a single protein.
Their framework, One Protein Is All You Need (OPIA), trains large language models directly on the mutational landscape of one protein using deep mutational scanning (DMS) data. From this, it learns structure, function and fitness without sequence databases or alignments.
Instead of learning from the internet, OPIA learns directly from the lab.
🔬 Applications and Insights
1️⃣ Single-protein learning
OPIA learns from the experimental mutations of one protein, capturing local rules of folding, binding and activity directly from measured data.
2️⃣ Accurate fitness prediction
Across ten benchmark proteins, OPIA matches or exceeds large-scale protein language models in predicting mutational fitness and stability, with strong correlation to experimental results.
3️⃣ Generalisation from local context
The model learns residue dependencies that mirror real structural and functional coupling, allowing it to predict unseen mutations accurately.
4️⃣ Data-efficient biology
OPIA demonstrates that deep learning can extract meaningful biological rules from single-protein experimental datasets, making modelling possible even when large databases do not exist.
💡 Why It’s Cool
OPIA flips the usual logic of protein modelling. Instead of training ever larger models on massive sequence corpora, it learns deeply from one molecule using experimental feedback. It offers a glimpse of lab-native AI, where models are grounded in real measurements rather than distant evolutionary proxies.
Sometimes, one protein really is all you need.
📄 Read the preprint
💻 Try the code
🧬 AlphaDIA: Deep Learning for Feature-Free, Transferable Proteomics
Data-independent acquisition (DIA) transformed proteomics throughput, but data analysis has lagged behind. Traditional pipelines rely on hand-tuned parameters and feature extraction, creating bottlenecks and limiting transfer between instruments.
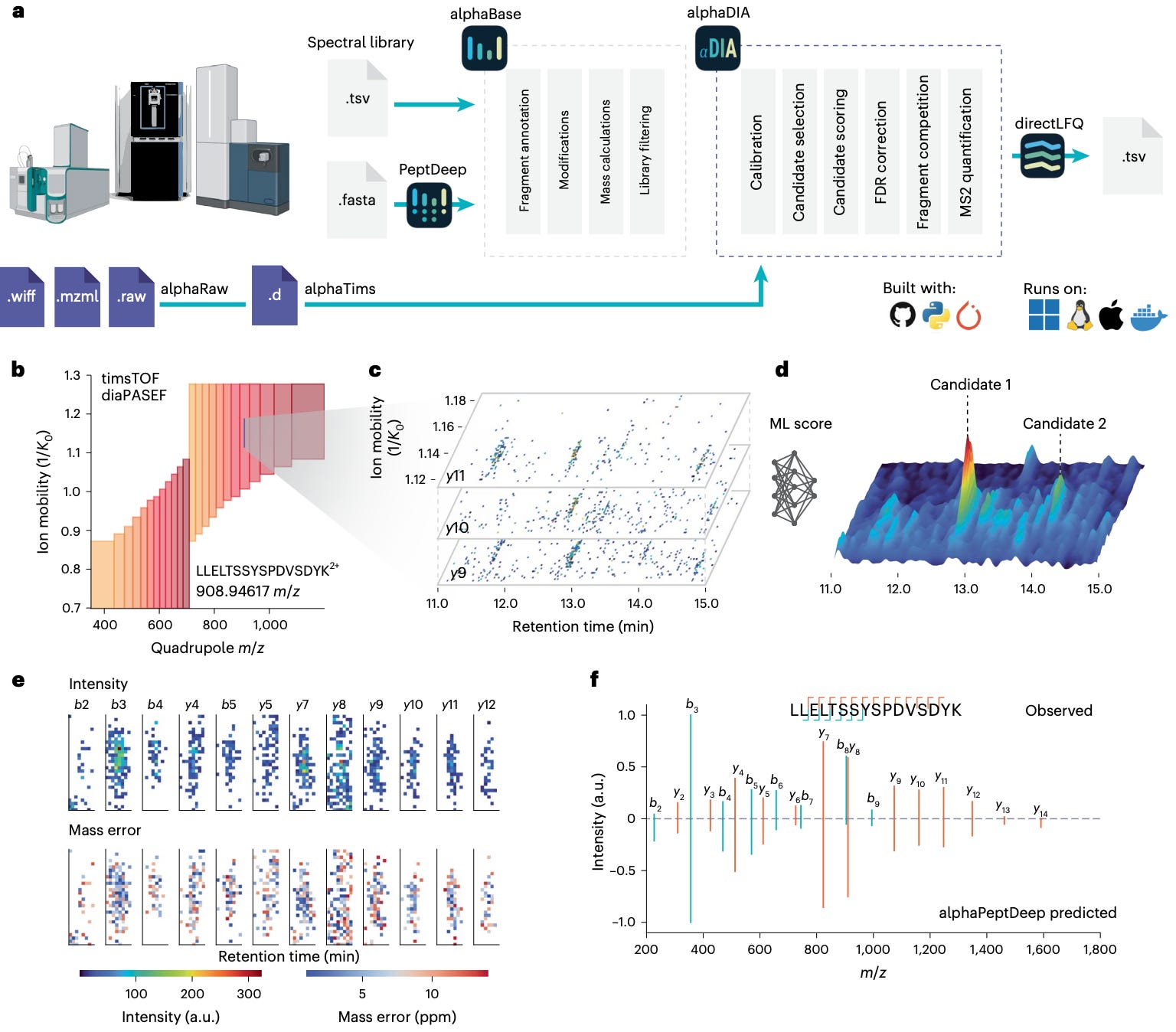
Researchers at the Max Planck Institute of Biochemistry and the University of Copenhagen built AlphaDIA, a deep-learning framework that reads DIA data directly from the raw signal.
No peak picking, no feature extraction — just direct interpretation of the detector output.
It connects raw ion traces to peptide identities through neural scoring and transfer learning, creating a faster, more accurate, and more adaptive DIA analysis pipeline.
🔬 Applications and Insights
1️⃣ Feature-free peptide identification
AlphaDIA interprets raw DIA signals directly, aggregating data across retention time, ion mobility, and m/z to identify peptides without predefined features.
2️⃣ Cross-platform generalization
Works seamlessly on timsTOF, Orbitrap Astral, and ZenoTOF data, adapting to different acquisition schemes through a unified learning model.
3️⃣ DIA transfer learning
Fine-tunes predicted spectral libraries from AlphaPeptDeep to each instrument and modification type, dramatically improving spectral correlation and retention-time prediction.
4️⃣ Discovery at scale
Identified 73,000 precursors and 6,800 protein groups in 21-minute HeLa runs and achieved a 48% gain in modified peptide detection using transfer learning.
💡 Why It’s Cool
AlphaDIA brings intelligence directly into the proteomics pipeline.
Instead of predefining what to look for, it learns patterns in the raw signal itself, then transfers that knowledge across datasets, instruments, and conditions.
It’s a foundation model for mass spectrometry: open, fast, and adaptive.
A system that learns from the data, not just processes it.
📄Check out the paper!
⚙️Try out the code.
Thanks for reading!
💬 Get involved
We’re always looking to grow our community. If you’d like to get involved, contribute ideas or share something you’re building, fill out this form or reach out to me directly.
Connect With Us
Have questions or suggestions? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website