
Foundation models for Biology: a short primer

Foundation Models in Biology represent one of the most important advances in bioinformatics in recent years. These models differ significantly from classical approaches in the field, as they are rooted in concepts originally developed in other artificial intelligence (AI) areas. As a result, even experienced bioinformaticians may find them challenging to understand. This article provides a summary of what you need to know about Foundation Models in Biology and their potential applications.
Many readers may be familiar with Large Language Models such as ChatGPT, which are trained on massive amounts of text data and are capable of generating responses that mimic human language. In a way, Foundation models for Biology try to accomplish similar objectives, but using the language of biology instead of human text.

When I first heard about Evo, a Foundation model based on Nucleotide data in Prokaryotes, I tried to chat with it, as if it was chatGPT. Its answer was “ACACACTGGA…” 😊
What are Foundation Models for Biology?
Generally speaking, Foundation Models in Biology are models trained on large quantities of biological data, such as genomic sequences or chemical structures, using architectures like the Transformer. This architecture is widely recognized for its role in training Large Language Models (LLMs) such as ChatGPT. The term “Foundation Models for Biology” has become popular thanks to the paper https://arxiv.org/abs/2402.04286 published in 2024.
Instead of focusing on text or human conversations, these models are trained on biological sequences or other types of biological data. By doing so, they learn the "language of life" by identifying and capturing hierarchical patterns inherent in the data.
For instance, a Foundation Model might be trained on gene expression data obtained from single-cell sequencing of blood tissue. Through this training, the model learns which genes are typically co-expressed in specific cell types. Remarkably, it uncovers these patterns and interactions purely from the data itself, without relying on prior knowledge of biological pathways or predefined gene sets.
What Are "Pre-training" and "Fine-tuning"?
Training a Foundation model is very expensive and requires large quantities of data. However, once a model has been pre-trained, it can be used and fine-tuned for other tasks. This means that we can take advantage of big atlases (provided the model is made public), and use them on smaller datasets, without having to download all the original data and train on it from scratch.
Pre-training: This initial phase involves training the foundation model on a vast dataset of biological information. For instance, the model may learn to predict the next nucleotide in a sequence or complete missing segments within it. Through this process, the model identifies and internalizes the fundamental patterns within the data, effectively building a comprehensive understanding of the "language of biology."
Fine-tuning: After pre-training, the model can be customized for specific tasks by adjusting its parameters and continuing training on smaller, task-specific datasets. This approach is advantageous because the model has already developed a foundational understanding of the data’s inherent patterns during pre-training, significantly reducing the resources needed for task-specific adaptation.
A pre-trained model might have learned general genomic patterns. By fine-tuning, it can be trained to classify patients as healthy or diseased based on their genomic data.
Example 1: using a pre-trained model to predict disease
Suppose you are developing a model to predict whether a patient is affected by a disease. You have access to a dataset of gene expression in cases/controls from a previous study.
A traditional bioinformatics approach might involve creating a data frame of gene expression values across all samples and training a model to differentiate between cases and controls. Popular methods include random forests, XGBoost, or logistic regression. In this setup, the predicted variable would be "disease," and the gene expression values would serve as inputs. Alternatively, you could perform a Differential Expression analysis to identify key genes or conduct a gene set enrichment analysis to determine which pathways are most affected.
However, these approaches often overlook the intricate relationships between genes. For example, Gene A might belong to the same pathway as Gene B, frequently being co-expressed. Gene C could act as a transcription factor for Gene D, but only in the presence of Gene E. Biology is incredibly complex, and many of these relationships remain poorly understood.
If your training dataset is sufficiently large, your model could potentially learn these gene relationships independently. You might also apply feature engineering to simplify the training process. Unfortunately, in most real-world scenarios, training datasets are far too small—often limited to cohorts of 10 or 20 samples, or even hundreds in the best-case scenario, which still might not suffice.
This is where Foundation Models come into play. By leveraging a pre-trained model trained on extensive gene expression datasets, such as the Gene Expression Atlas, you can transform your small dataset into embeddings. These embeddings encapsulate the knowledge of gene interactions and relationships acquired during pre-training. By using these embeddings instead of raw gene expression values, your predictions become more accurate and biologically informed, integrating the wealth of insights the Foundation Model has learned.
Example 2: Imputation and Upscaling of Microarrays
I like this example from the CpGPT paper https://www.biorxiv.org/content/10.1101/2024.10.24.619766v1.full, which presents a model trained on Methylation data.
Despite the rise of Next-Generation Sequencing (NGS) technologies, microarrays remain one of the most cost-effective methods for profiling methylation samples. A significant amount of data has been generated using older Illumina microarray technologies, such as the HumanMethylation BeadChips, which profile only 27K or 450K CpG sites. Vast repositories of such data exist in PubMed publications and GEO datasets. In contrast, modern arrays can genotype over 900K sites, leaving the older datasets lacking critical information.
Is there a way to use the data from all these old chips, and “update it” to the newest one, predicting the sites that were not present in the original assay? The answer, proposed in the CpGPT paper, is yes. They trained a transformer model on methylation data from many sources, including old and new chips. The model learned how to impute new sites and predict the genotype of the missing CpGs based on data. To validate the results, they used the imputed datasets to predict age, and obtained much lower errors.

Excerpt from Figure 3 from the CpGPT paper (https://www.biorxiv.org/content/10.1101/2024.10.24.619766v1.full) showing better performances when predicting age using data reconstructed with CpGPT. Notice, for example, the lower Mean Absolute Error (MAE) scores for the reconstructed data.
Similarly, a foundation model can impute missing SNPs from Genotype data and other data types. Foundation models can also be used for generating new datasets synthetically - the preciousGPT3 paper (https://www.biorxiv.org/content/10.1101/2024.07.25.605062v1describes this neatly.
Example 3: Predicting Gene Expression from ChIP-Seq data
The latest Foundation Model published, at the time of writing this article, is Generalized Expression Transformer (GET) https://www.nature.com/articles/s41586-024-08391-z. It is trained on chromatin accessibility and gene expression data.
Let’s assume you have access to a ChIP-Seq dataset, and you want to predict whether a specific gene is expressed; this is not a trivial task. Traditional methods for predicting gene expression from chromatin accessibility often rely on statistical or machine-learning approaches. One common method is to identify accessible regions near gene promoters and correlate these regions with expression levels using linear regression or other basic predictive models. Alternatively, tools like ChromHMM and ATAC-seq pipelines can integrate chromatin states and transcription factor motifs to make predictions. While these methods can provide valuable insights, they typically require extensive feature engineering and may not capture the full complexity of regulatory interactions.
GET bypasses these limitations by leveraging a Transformer architecture. By training on vast datasets, it learns intricate patterns between chromatin accessibility and gene expression, capturing interactions across the genome. This approach enables highly accurate predictions, even for genes with complex regulatory landscapes, demonstrating the potential of Foundation Models to revolutionize our understanding of gene regulation.
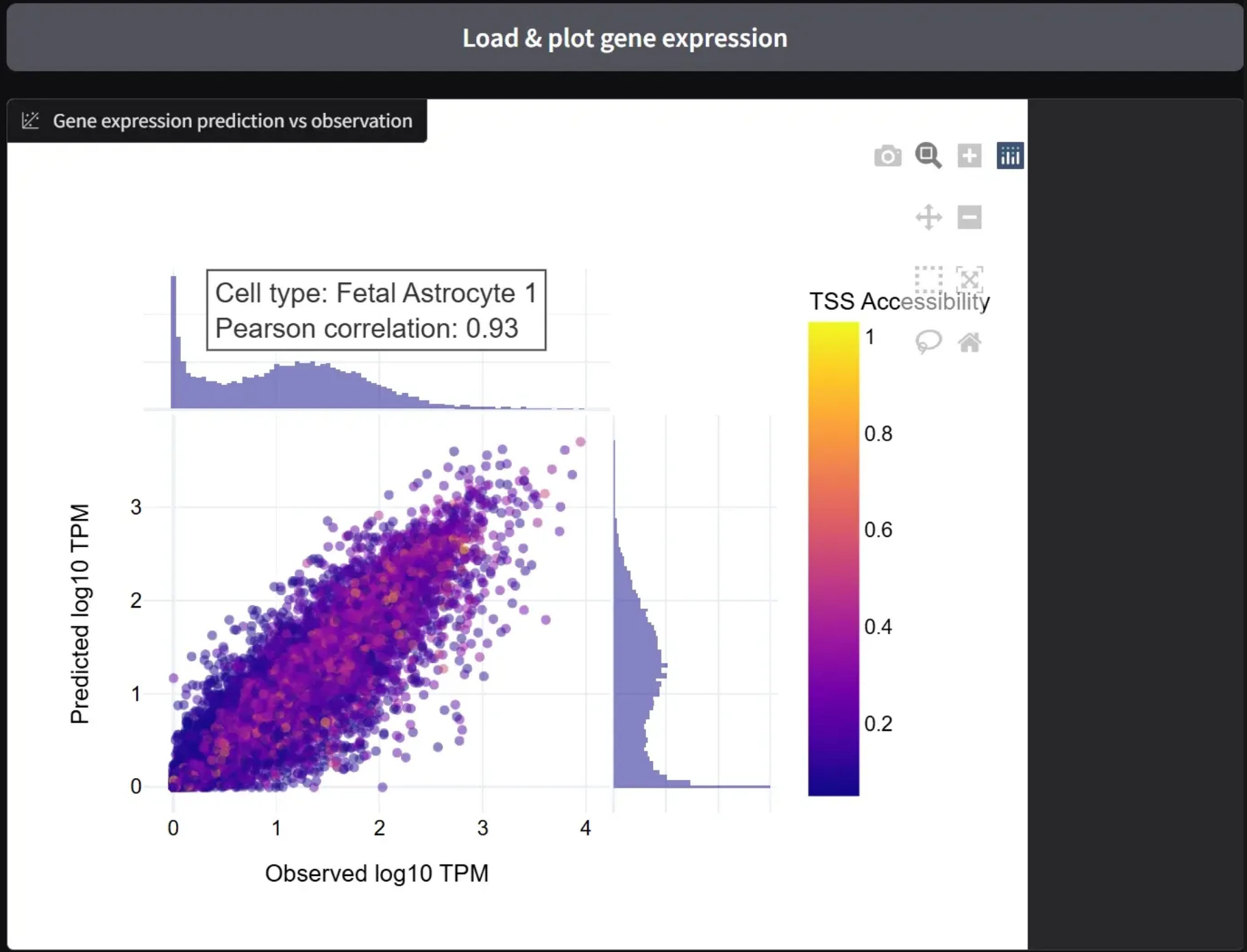
Predicted Gene Expression for Fetal Astrocyte cell types, using the GET model from its HuggingFace page. The model takes a matrix of DNA accessibility patterns and predicts gene expression. The Pearson correlation of 0.93 for this example shows a good correlation between predicted data and the gold standard.
Some Key concepts related to Transformers
Let’s dive into some technical details. Foundation models are based on concepts derived from other areas of AI, and some of the terms may be complex to understand, even for people who have been in bioinformatics for many years. I remember reading the Evo paper for the first time and struggling to understand what was happening.
Most Foundation models in biology are based on the Transformer architecture. Although there are many exceptions (e.g., EVO is based on StrypedHyena, a different architecture), understanding the basics of how Transformers work can help you get started.
Encoder and Decoder
The Transformer architecture is composed of two components: Encoders and Decoders.
The Encoder takes an input and translates it into a series of numerical vectors called embeddings. The input depends on what is being modeled, but it can be nucleotides, proteins, gene expression values, genetic data, or a combination of these. Examples of Encoders include DNA BERT (https://academic.oup.com/bioinformatics/article/37/15/2112/6128680), scBERT (https://www.nature.com/articles/s42256-022-00534-z), and others.
The Decoder takes the embeddings and uses them to perform generative tasks, such as generating new data or imputing missing data. CpGPT, cited previously in this paper, is an example of a Decoder.
Embeddings:
One of the key concepts for understanding Foundation Models is that of embeddings.
Deep Learning algorithms cannot process text or categorical data directly. In natural language processing, text is transformed into vectors of numbers that represent words and their positions in a sentence. When a Decoder is trained, the model learns to transform these numbers in a way that reflects not only the word’s position but also its meaning and relationship to other words.
Similarly, in biological data, nucleotide sequences or gene names are transformed into numerical embeddings. These embeddings encode both the identity and context of each element in the data. For example, in a nucleotide sequence, embeddings might capture the base type, genomic position, and neighbouring elements.
Designing effective embeddings requires selecting features relevant to the task. For instance:
Evo uses embeddings optimized for evolutionary data.
scGPT encodes single-cell RNA-seq data, including metadata like cell type and experimental batch.
CpGPT incorporates features such as CpG methylation status, genomic position, and nearby base context.
Attention Mechanism:
In natural language, the attention mechanism models how words in a sentence relate to one another. For example, it helps associate adjectives with their corresponding nouns or distinguish words with multiple meanings depending on context.
In biological data, the attention mechanism models relationships between elements such as regulatory sequences, pathways, and gene interactions. This allows the model to learn complex dependencies within the data.
Generative Training:
When training a Decoder, the model is trained to predict the next word in a sentence. For example, given the input sentence “The black cat is on,” the model predicts the next word.
To optimize training, the model uses all elements of the sentence as training data. For example, with the input sentence “The black cat is on the table,” the model makes predictions starting with “The” to predict “black,” then with “The black” to predict “cat,” and so on.
This method enables parallelized training on GPUs, making it highly efficient.
In genomic data, Decoders can predict every nucleotide based on its preceding sequence. While genomics data isn’t inherently sequential, this approach is still widely used and is an area of active research.
Encoders, on the other hand, are trained differently. Instead of predicting the next token, they predict masked or missing data, learning to understand the context and fill gaps in the information.
What’s Next for Foundation Models in Biology?
2024 has seen the publication of many Foundation Models for Biology. Some of these models have been released publicly, while others were trained by private companies.
Hopefully, this article helped summarise these models' key concepts and applications. We live in exciting times, as Foundation Models are transforming how we approach biological problems. They will make it easier to capitalize on the massive amounts of data generated by public consortia, especially if these models are made publicly available with their weights. For good or bad, they are going to become an integral part of Bioinformatics in the near future, and they will soon be part of the standard curriculum for anybody working in the field.