
Glasgow’s ExplainBind, UIUC’s 4D Whole-Cell Model, and Cambridge’s MACE-POLAR-1
Kiin Bio's Weekly Insights

Welcome back to your weekly dose of AI news for Life Science!
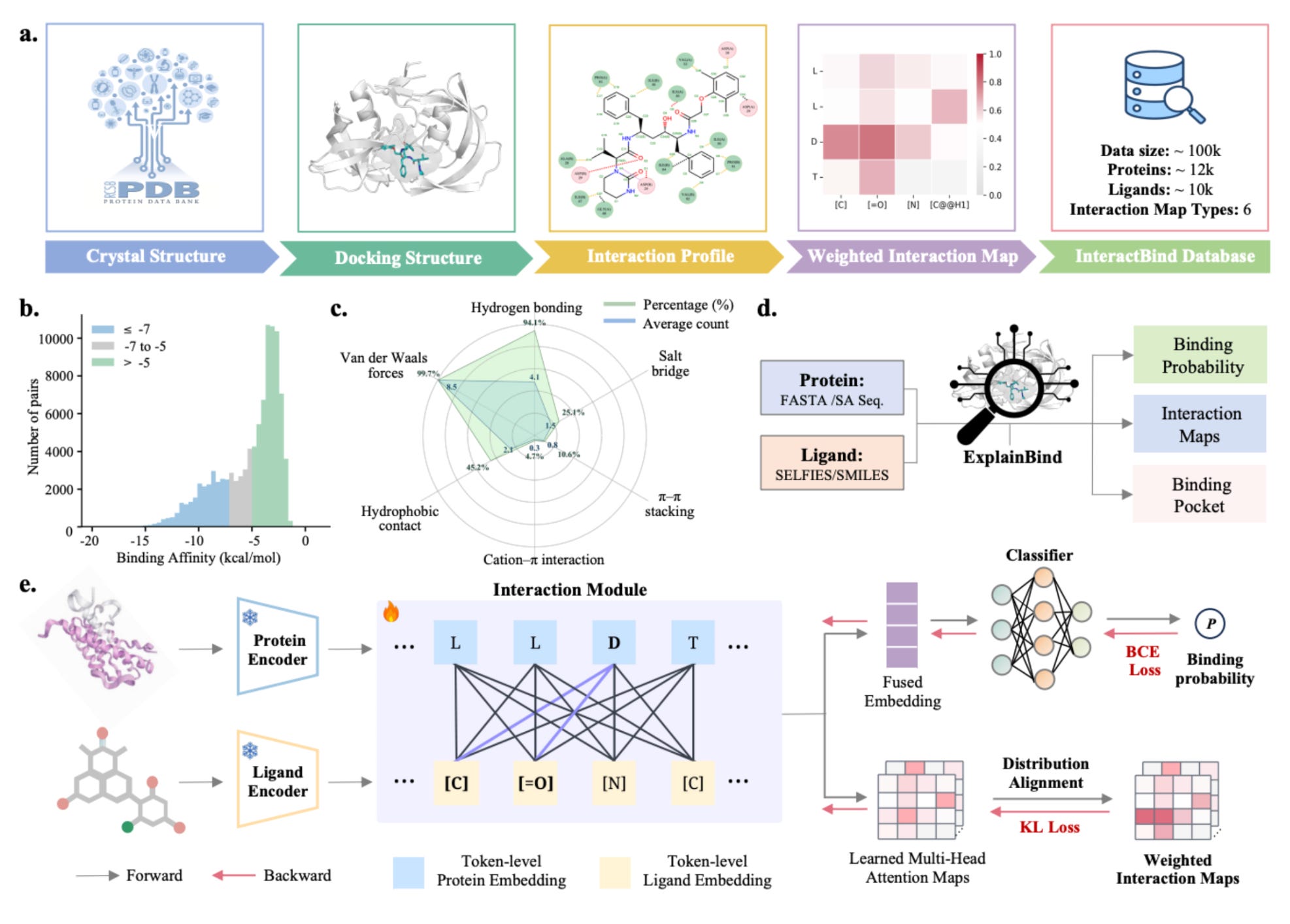
ExplainBind: Interpretable protein-ligand binding prediction grounded in non-covalent interactions
🔬 Protein-ligand binding sits at the heart of drug discovery. Most ML models treat it as a pattern-matching problem, offering predictions without chemical insight. You get a score, not a mechanism.
ExplainBind changes that. Developed by researchers at the University of Glasgow, the Broad Institute, and Harvard, it predicts binding by explicitly modelling the non-covalent interactions (hydrogen bonds, hydrophobic contacts, electrostatic forces, van der Waals) that drive molecular recognition.
🧬 The model supervises token-level cross-attention using non-covalent interaction maps derived from PDB complexes, linking predictions to physicochemical forces at play. It operates from protein sequences and ligand representations, with no 3D structure required.
⚡ Evaluated on Angiotensin-Converting Enzyme and L-2-hydroxyglutarate dehydrogenase, ExplainBind achieved an AUROC of 0.993, outperforming eight baseline models, and enriched potent ACE inhibitors at the top of virtual screening rankings.
🎯 Applications & Insights
1️⃣ Interpretable Binding Prediction
AUROC of 0.993 against eight baselines, grounded in physicochemical interaction modelling rather than statistical correlation. Applies to proteins without solved 3D structures.
2️⃣ Binding Pocket Localisation from Sequence
Infers binding pockets from sequence data alone by analysing attention patterns between residues and ligand atoms. No crystal structure required.
3️⃣ Retrospective Ligand Prioritisation
ACE inhibitors enriched at the top of virtual screening rankings, demonstrating practical utility for filtering large chemical libraries.
4️⃣ Residue-Level Mechanistic Insight
Attention mechanisms map amino acid residues to ligand atoms in patterns corresponding to real non-covalent interactions, revealing which residues drive binding.
💡 Why This Is Cool
Interpretability in drug discovery usually means post-hoc explanations bolted onto black-box models. ExplainBind builds the chemistry in from the start. Supervising attention with non-covalent interaction maps is a cleaner approach, and the residue-level outputs are useful for medicinal chemists, not just reviewers.
📖 Read the paper
💻 Try the code
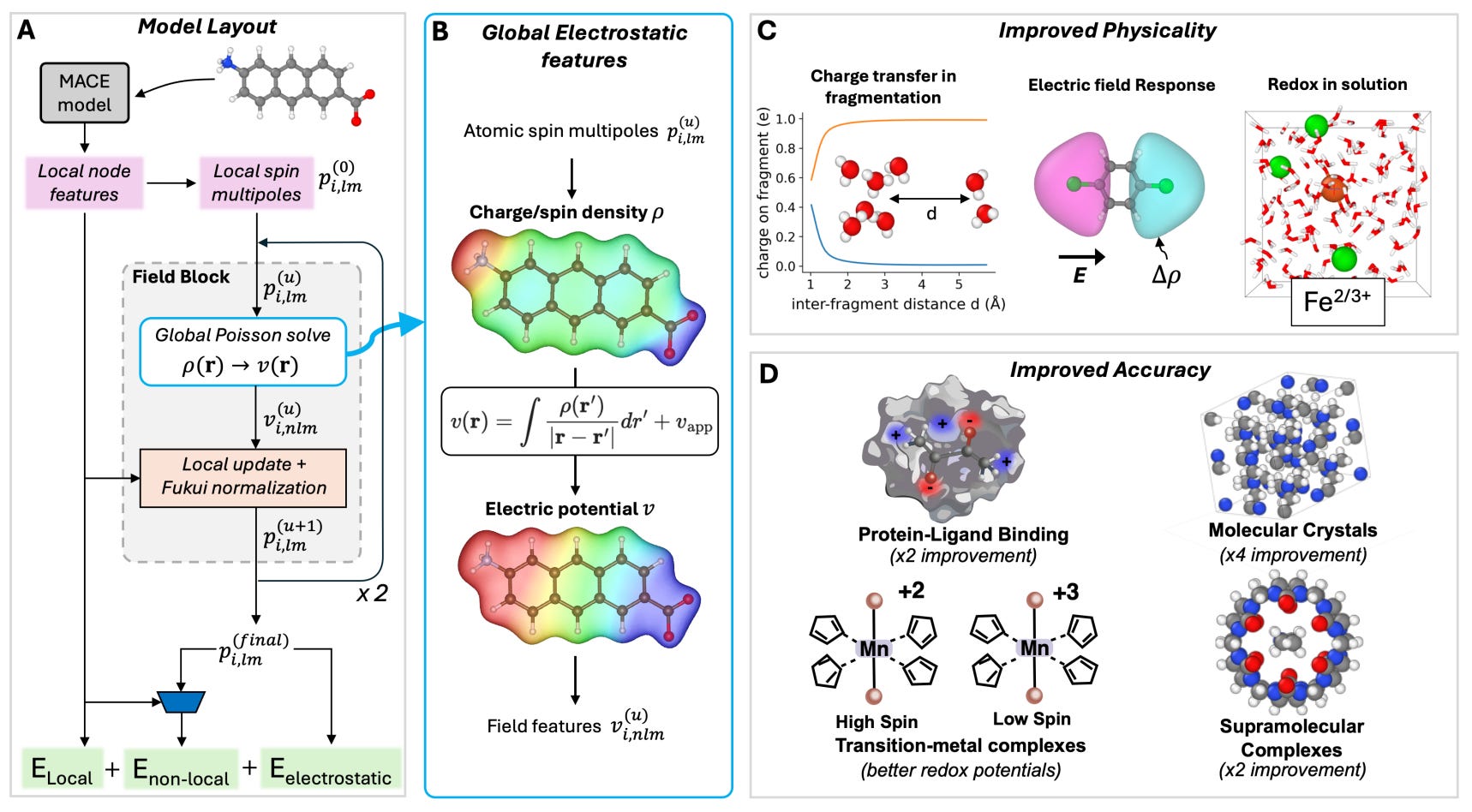
MACE-POLAR-1: A polarisable electrostatic foundation model for molecular chemistry
🔬 Short-range ML potentials miss the long-range electrostatic effects governing solvation, protein-ligand binding, and transition metal chemistry. Standard MLIPs treat charges as fixed, breaking down wherever electron density redistribution matters.
MACE-POLAR-1 extends the MACE architecture with explicit polarisation and charge equilibration, trained on OMol25: 100M hybrid DFT calculations.
🧬 A non-SCF induction formalism and learnable Fukui functions handle charge/spin equilibration, decomposing energy into local, non-local, and electrostatic terms across variable charges, spins, and ions.
⚡ Results: fourfold improvement on protein-ligand interactions (PLF547, MAE 0.37 kcal/mol), sub-kcal/mol crystal lattice energies, and accurate liquid water density from 270-330 K.
⚗️ Applications & Insights
1️⃣ Protein-Ligand Binding
Fourfold improvement over short-range baselines on PLF547, capturing long-range electrostatics critical to binding affinity.
2️⃣ Solvation and Transition Metal Chemistry
Accurate solvation structure of Fe ions, redox chemistry, and ionisation energies of hydrated transition metals.
3️⃣ Molecular Crystals
Sub-kcal/mol lattice energy errors on X23-DMC, where long-range electrostatics govern crystal packing.
4️⃣ Electric Field Response
Correctly models applied electric fields, enabling electrochemical and polarisation-sensitive applications.
💡 Why This Is Cool
Foundation models for chemistry have largely ignored polarisation. MACE-POLAR-1 puts it at the centre, delivering chemical accuracy across thermochemistry, condensed matter, and biomolecular systems.
📖 Read the paper
💻 Try the code
🤖 Get the model
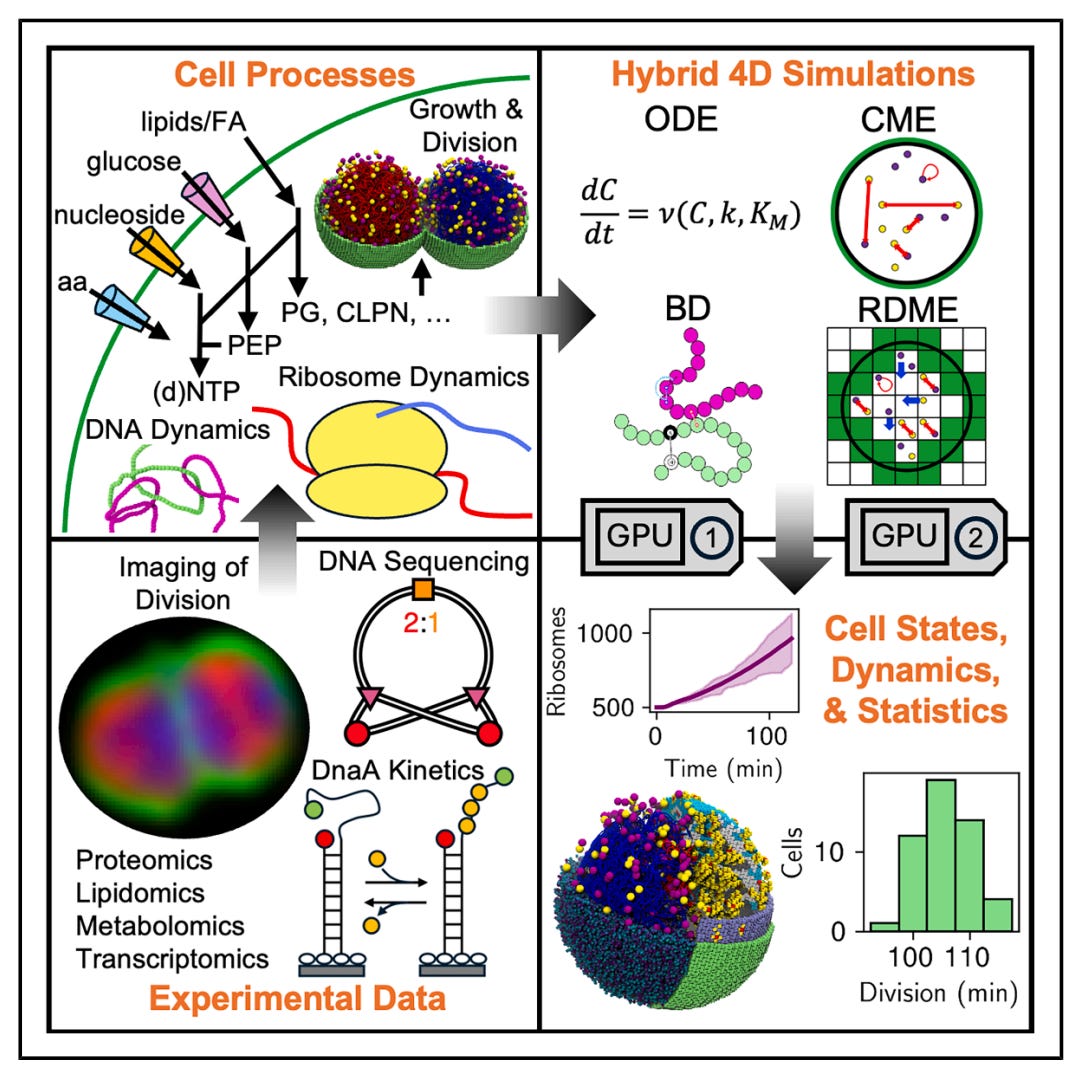
Bringing the Genetically Minimal Cell to Life on a Computer in 4D
🔬 JCVI-syn3A, the genetically minimal bacterium with just 493 genes, is the simplest living organism. Researchers at UIUC, Johns Hopkins, Harvard, and the J. Craig Venter Institute built the first 4D whole-cell model (4DWCM) of its ~100-minute cell cycle.
🧬 Three simulation engines run coupled: RDME for stochastic gene expression, CME/FBA for metabolic kinetics, and Brownian dynamics for chromosome replication and segregation.
⚡ 50 replicate simulations recover the ~105-minute doubling time, ribosome counts, mRNA distributions, and spatial heterogeneity, validated against sequencing, proteomics, and imaging. Each cell is unique.
🔬 Applications & Insights
1️⃣ First Complete 4D Cell Cycle Simulation
All 493 genes, full metabolism, chromosome replication, ribosome biogenesis, and division modelled in space and time.
2️⃣ Hybrid Multi-Scale Framework
RDME, CME/FBA, and Brownian dynamics fully coupled: a blueprint for whole-cell computational biology.
3️⃣ Stochastic Heterogeneity Across Replicates
50 simulated cells show unique molecular partitioning at division, predicting variability bulk experiments cannot resolve.
4️⃣ Multi-Omics Validated
Ribosome counts, mRNA half-lives, protein distributions, and division timing match sequencing, proteomics, imaging, and cryo-ET data.
💡 Why This Is Cool
Whole-cell modelling has been an aspiration for decades. This is the closest any model has come to a complete, spatially resolved simulation of life.
📖 Read the paper
💻 Try the code
🗓️ Events & Competitions
The best competitions, hackathons, and community challenges in AI x life sciences, curated weekly. Know something worth featuring? Reply and let us know.
Recap of Berlin Bio AI Hackathon (Feb 27-28) with Hannah Payette Peterson, one of the organisers:
Last weekend, 76 researchers from across Europe gathered in Berlin for a 24-hour biotech x AI hackathon co-organised by Nucleate Germany, JUNI, and Project Europe. Three challenge tracks ran in parallel: Protein Design with Adaptyv Bio 🧪, Genome Modelling and Synthesis with Serova 🔬, and Agentic AI in Life Sciences with Google DeepMind 💡
The overall winner, Omnomnomics, was a team that formed on day one from five strangers across different countries 🌍 The Agentic AI track winner, Delta Wave x NanoSpec, built an agent to classify longevity research evidence using Gemini and DeepMind’s MedGema ⚡ A Stockholm edition is already being planned for later this year, watch out for more details on this. 🇸🇪
More upcoming events:
BioHackathon Edinburgh 2026 | March 20-22, Edinburgh
Three days at the University of Edinburgh bringing together life scientists, programmers, and industry partners to hack on real biological challenges. Tracks cover academic research (gene regulation, drug discovery, imaging), industry innovation, and a non-coder track for experimental design and project management. Applications are closed, but one to watch if you’re at a UK university for next year.
BioHackathon Europe 2026 | November 9-13, Barcelona
ELIXIR’s annual international bioinformatics hackathon, running since 2018. 160+ participants, five days of collaborative coding on open bioinformatics infrastructure and tools. The call for project proposals opens March 16 and closes April 15 - so if you want to lead a project, that’s your window.
Thanks for reading!
💬 Get involved
We’re always looking to grow our community. If you’d like to get involved, contribute ideas or share something you’re building, fill out this form or reach out to me directly.
Connect With Us
Have questions or suggestions? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website