
Helmholtz's RegVelo, Calico's TTM, and NIH's Path2Space
Kiin Bio's Weekly Insights

Welcome back to your weekly dose of AI news for Life Science!
Keeping up with AI x life science news can get exhausting.
It’s scattered across LinkedIn, X, Substack, arXiv, Slack, newsletters... and you still somehow miss the things that actually matter. Too much noise, not enough signal.
We’re building something to fix that: a smarter, more powerful way to stay on top of what’s actually relevant to you.
But we want to build it with you, not just for you. Take 2 minutes to tell us what’s missing. What you share will directly shape what we build, and you’ll be the first to benefit from it.
🇺🇸 We’re heading to Bio-IT World in Boston, May 19-21.
Our CEO Filippo and CTO Bogdan will be there and would love to meet anyone thinking about:
How AI is actually changing preclinical workflows (not just the hype)
Why drug discovery is a systems problem, not just a science one
What it takes to go from 5-year timelines to something radically faster
No pitch, just good conversation. If any of that’s on your mind, reach out - we’ll find a time to grab a coffee.
RegVelo: Gene-Regulatory-Informed Dynamics of Single Cells
🔬 RNA velocity models cellular dynamics but ignores gene regulatory interactions. Conversely, gene regulatory network inference methods neglect dynamics entirely. No existing approach jointly captures both, limiting our ability to simulate perturbations and predict how regulatory changes drive cell fate decisions.
RegVelo from Helmholtz Munich and Fabian Theis’s lab bridges this gap. It is an end-to-end deep generative framework that jointly infers transcriptome-wide splicing kinetics and gene regulatory interactions from scRNA-seq data, producing an actionable in silico cell for perturbation simulation.
🧬 RegVelo encodes unspliced and spliced RNA into a latent space, then models transcription as a regulated process governed by a GRN weight matrix. A parallel high-dimensional ODE solver couples all gene dynamics simultaneously rather than treating genes independently. Prior GRN knowledge from ATAC-seq or public databases constrains the network, while data-driven refinement learns new regulatory edges and edge weights.
⚡ On cell cycle data, RegVelo achieves a cross-boundary correctness of 0.864, velocity consistency of 0.873, and significantly outperforms scVelo and veloVI (p < 0.001). For GRN inference, it ranks first among six methods on edge prediction (median AUROC = 0.59) and achieves AUROC = 0.95 for identifying known lineage driver TFs across four hematopoietic lineages. Predictions validated by CRISPR-Cas9 knockout and single-cell Perturb-seq in zebrafish neural crest.
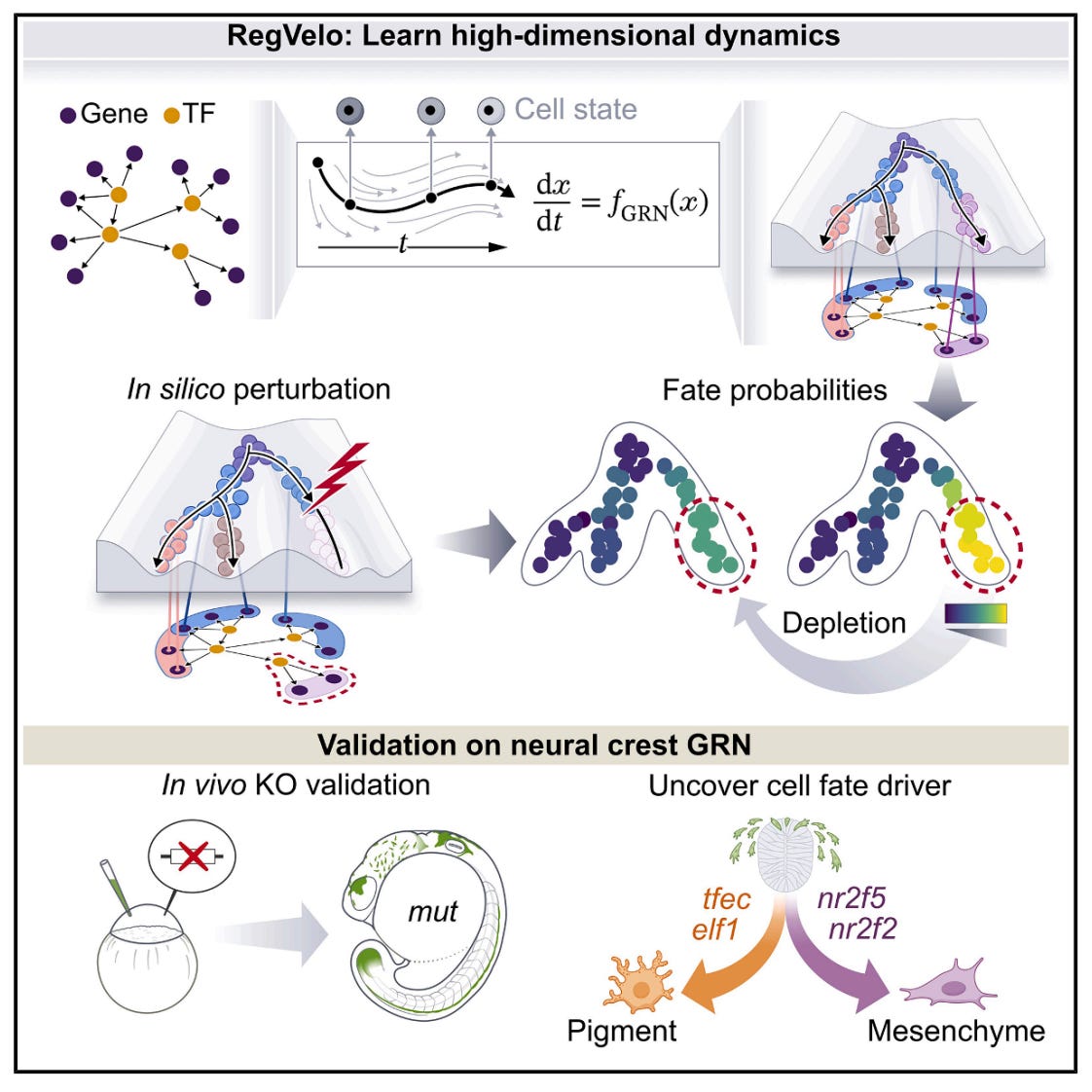
🔬 Applications and Insights
1️⃣ In Silico Perturbation Screening
Masking regulons and comparing velocity fields lets researchers simulate gene knockouts computationally, predicting cell fate shifts before running wet-lab experiments.
2️⃣ Cell Fate Driver Discovery
Applied to zebrafish neural crest, RegVelo identified tfec as a key early driver and elf1 as a regulator of pigment cell fate, both validated in vivo with CRISPR-Cas9.
3️⃣ Lineage-Specific GRN Recovery
In hematopoiesis, RegVelo recovered known lineage drivers (Smarca1, Pdx1, Mnx1, Hhex) across four lineages with high ranking accuracy (AUROC = 0.95).
4️⃣ Uncertainty-Aware Predictions
As a Bayesian generative model, RegVelo quantifies intrinsic and extrinsic cell state uncertainty, giving confidence estimates for both velocities and inferred regulatory edges.
💡 Why This Is Cool
This is the first framework to couple RNA velocity with gene regulatory networks in a single generative model. Rather than inferring dynamics and regulation separately and hoping they align, RegVelo learns them jointly. The result is a model that can simulate what happens when you perturb the regulatory wiring, with predictions validated from in silico all the way to in vivo knockouts. That closes the loop from computational hypothesis to experimental confirmation.
📄 Read the paper
💻 Try the code
TTM: Triplet Tumbling Microscopy Enables In Situ Quantification of Protein Complex Assembly and Dynamics
🔬 Protein-protein interactions drive nearly every cellular process, but measuring them inside living cells remains limited. FRET requires two labels and prior knowledge of interacting partners, while fluorescence anisotropy only works for small proteins below 50 kDa. There is no broadly applicable way to quantify protein complex size and binding dynamics in situ in real time.
TTM (Triplet Tumbling Microscopy) from Calico Life Sciences solves this by measuring rotational diffusion of protein complexes using only a single fluorescent tag. By leveraging long-lived triplet states in fluorescent proteins, TTM extends the measurable timescale from nanoseconds to hundreds of microseconds, covering the full range of cellular protein complexes.
🧬 TTM uses a pulsed excitation sequence: a 488 nm pulse generates triplet states aligned with the excitation polarisation, then an infrared trigger pulse (785-940 nm) reads out their orientation after a variable delay. As proteins tumble, the triplets lose alignment at a rate proportional to complex size. Rigid fluorescent protein tags (truncated mVenus and mStayGold) ensure tag motion faithfully reports target motion.
⚡ In purified protein systems, tumbling time constants scale linearly with molecular weight (r squared = 0.99). In living U2OS cells, TTM resolves complexes from 41 to 195 kDa from single-cell recordings (r squared = 0.85). It detects the approximately 10% size change from E6AP binding HPV16 E6 protein, quantifies p53 homo-oligomerisation states across nine point mutations, and tracks rapamycin-induced dimerisation dynamics in real time at approximately 3 Hz.
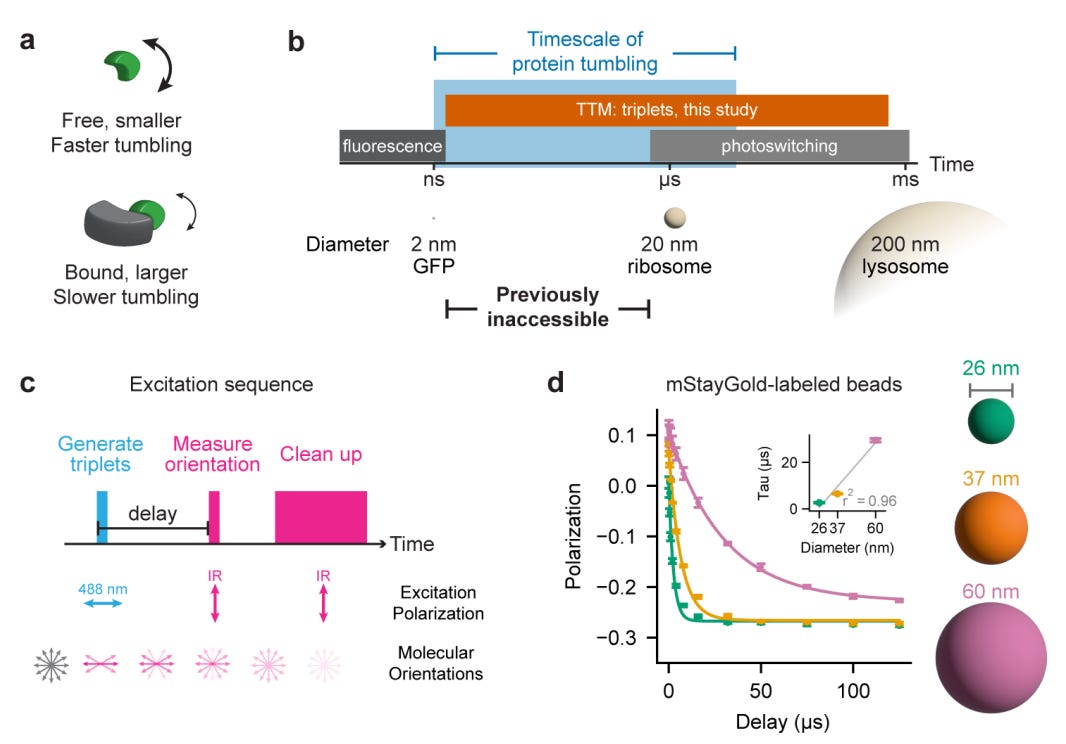
🔬 Applications and Insights
1️⃣ Single-Tag Interaction Detection
Unlike FRET, TTM requires only one fluorescent label and no prior knowledge of binding partners, making it applicable to uncharacterised or unexpected interactions.
2️⃣ Live-Cell Binding Dynamics
Real-time imaging at approximately 3 Hz captures the kinetics of complex formation as it happens, not just endpoint measurements.
3️⃣ Oligomerisation State Profiling
TTM distinguishes monomers, dimers, and tetramers of p53 in cells, revealing how tetramerisation domain mutations shift the equilibrium between functional states.
4️⃣ Standard Microscope Compatibility
The hardware requirements (pulsed lasers and an intensified camera) are compatible with most fluorescence microscopes, lowering the barrier to adoption across labs.
💡 Why This Is Cool
TTM fills a gap that has persisted for decades in cell biology: measuring how big a protein complex is inside a living cell, in real time, with a single label. The ability to track binding dynamics and oligomerisation states at physiological concentrations opens the door to studying protein interactions in their native context rather than in lysates or reconstituted systems. One tag, one measurement, real answers.
📄 Read the paper
Path2Space: AI-Predicted Spatial Transcriptomics Unlocks Breast Cancer Biomarkers from Pathology
🔬 Spatial transcriptomics is transforming our understanding of tumour heterogeneity, but its high cost limits large-scale biomarker discovery. Previous efforts to predict gene expression from histopathology slides have been restricted to small gene sets, precluding survival and treatment response analyses in large clinical cohorts.
Path2Space from NIH’s National Cancer Institute and Cedars-Sinai predicts the spatial expression of thousands of genes directly from routine H&E-stained histopathology slides. Trained on extensive breast cancer spatial transcriptomics data, it outperforms 21 established methods and enables scalable biomarker discovery without molecular assays.
🧬 Path2Space uses CTransPath, a digital pathology foundation model, to extract features from colour-normalised tile images around each spatial transcriptomics spot. A multilayer perceptron predicts log-transformed expression for 14,068 genes per spot. A spatial smoothing step averages predictions with neighbouring spots to mitigate technical variability. Trained on the Bassiouni et al. cohort comprising 56,567 matched image-expression spot pairs from 14 patients.
⚡ Median gene-wise PCC of 0.38 (smoothed) across 14,068 genes, with 6,629 genes exceeding PCC > 0.4. Binary classification of high versus low expression yields median AUC of 0.70, with 3,116 genes surpassing 0.75. Generalises robustly across three independent external cohorts (HEST, Martinez, HTAN). Applied to 976 TCGA breast cancer patients, Path2Space identifies three prognostic SpatioTypes and predicts chemotherapy and trastuzumab response at accuracy levels equal to or exceeding bulk sequencing biomarkers.
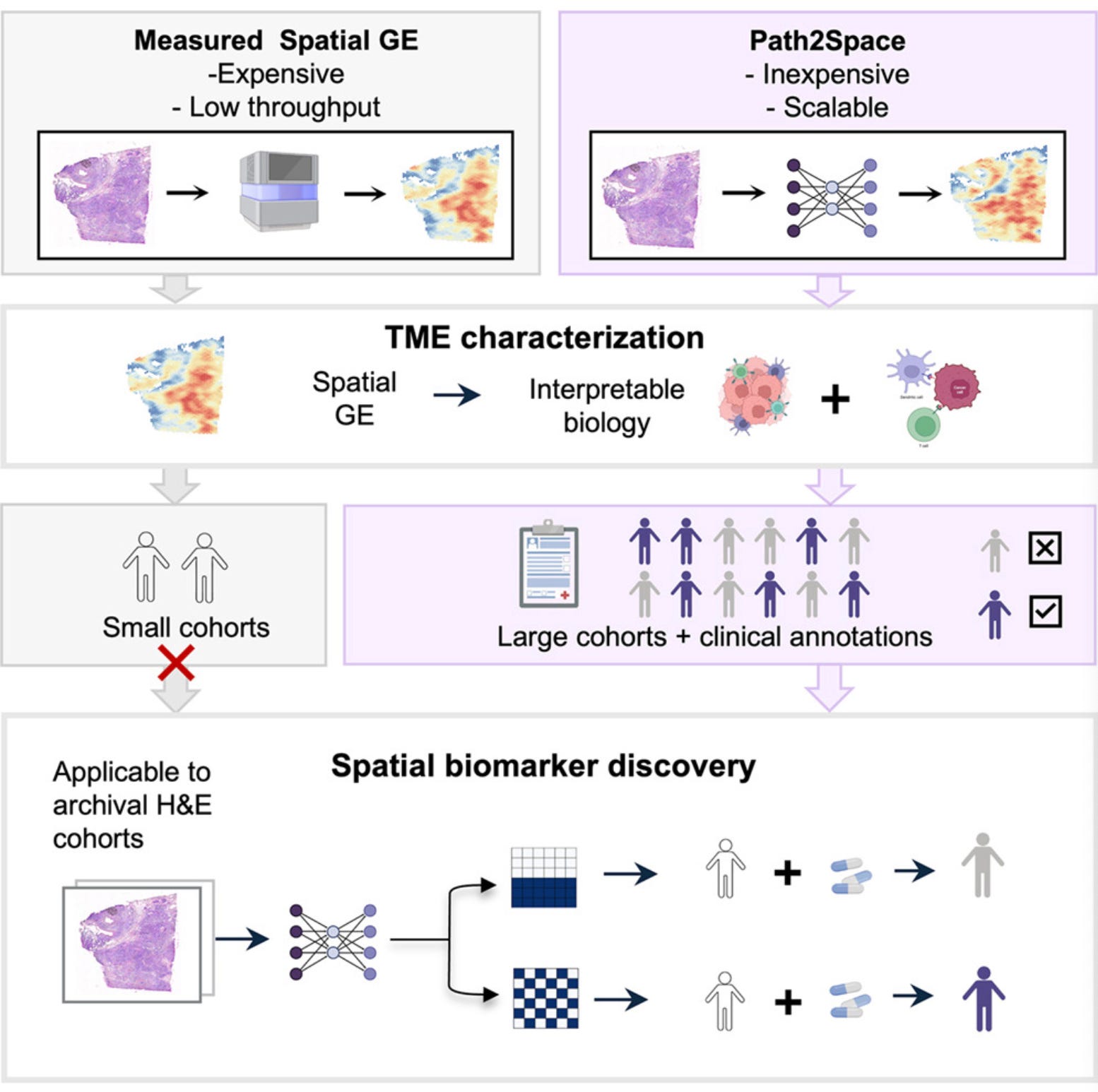
🔬 Applications and Insights
1️⃣ Low-Cost Spatial Biomarker Discovery
Derives spatial gene expression landscapes from routine pathology slides without expensive molecular assays, enabling large-cohort studies previously limited by cost.
2️⃣ Prognostic Breast Cancer Subtyping
Unsupervised clustering of predicted spatial transcriptomic profiles identifies three SpatioTypes with distinct biology and survival outcomes across 976 patients.
3️⃣ Treatment Response Prediction
Spatial biomarkers from H&E slides predict response to chemotherapy and trastuzumab at accuracy levels matching or exceeding those from bulk tumour sequencing.
4️⃣ Archival Tissue Applicability
Works on standard FFPE and fresh-frozen archival tissue, meaning existing hospital slide collections can be retrospectively analysed without new sample collection.
💡 Why This Is Cool
Spatial transcriptomics has been too expensive to run on the thousands of patients needed for robust biomarker discovery. Path2Space sidesteps this entirely by inferring spatial gene expression from H&E slides that hospitals already collect for every tumour. Turning routine pathology into a spatial omics readout could democratise precision oncology for any institution with a slide scanner.
📄 Read the paper
💻 Try the code
🗓️ Events & Competitions
The best competitions, hackathons, and community challenges in AI x life sciences, curated weekly. Know something worth featuring? Reply and let us know.
More upcoming events:
London Protein Design Day | June 23, Imperial College London
The first edition of a one-day symposium bringing together London’s protein design community and beyond. Programme spans AI-driven design, molecular dynamics, and bioinformatics, with applications across enzymes, antibodies, and materials. Organised by Pietro Sormanni, Rebecca Birolo, and Jakub Lála. Abstract deadline for poster/oral presentations is this Saturday (May 17). In person only.
BioHackathon Europe 2026 | November 9-13, Barcelona
ELIXIR’s annual international bioinformatics hackathon, running since 2018. 160+ participants, five days of collaborative coding on open bioinformatics infrastructure and tools. The call for project proposals opens March 16 and closes April 15 - so if you want to lead a project, that’s your window.
Thanks for reading!
💬 Get involved
We’re always looking to grow our community. If you’d like to get involved, contribute ideas or share something you’re building, fill out this form or reach out to me directly.
Connect With Us
Have questions or suggestions? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website