
Institut Pasteur's scPRINT2, EPFL’s DynaMate, and Harvard’s PROTON
Kiin Bio's Weekly Insights

Merry Christmas and welcome back to your weekly dose of AI news for life science!
What’s your biggest time sink in the drug discovery process?
🧬 scPRINT2: A Foundation Model That Finally Generalises Across Single-Cell Data
What if we stopped retraining models every time a new single-cell dataset showed up?
Anyone working with single-cell RNA-seq knows the cycle. New tissue, new protocol, new batch effects, and suddenly your model no longer holds up. Most approaches are trained on a curated slice of data and struggle the moment things get messy.
scPRINT2, developed by the team at Institut Pasteur, Université Paris Cité and CNRS, takes a different approach. It is a pretrained single-cell foundation model built from 43 million cells across more than 1,100 datasets, deliberately spanning diverse tissues, protocols and noise profiles. Instead of avoiding the mess, scPRINT2 learns from it.
The goal is simple: generalisation that actually holds up in the wild.
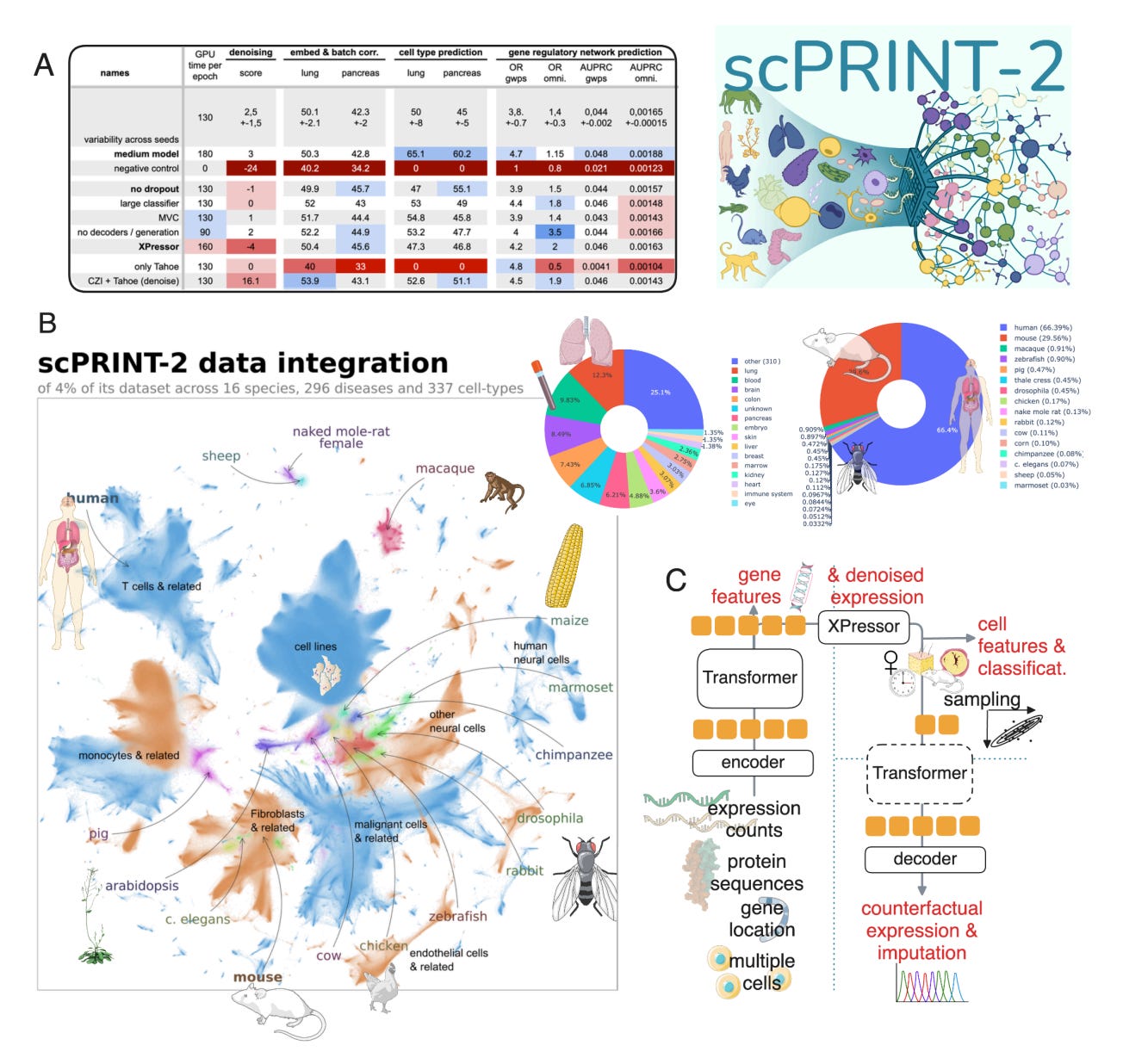
🔬 Applications and Insights
1️⃣ Zero-shot cell type prediction that holds up
Across six held-out datasets, scPRINT2 outperformed scVI and scANVI by 13.6 per cent and 10.7 per cent without any retraining. That level of zero-shot performance is rare for models at this scale.
2️⃣ Robust to missing genes
Even when up to 90 per cent of genes were masked, scPRINT2 maintained strong performance. This is particularly relevant for sparse datasets and panel-based assays where full transcriptomes are not available.
3️⃣ Gene expression imputation that makes biological sense
On held-out test sets, scPRINT2 outperformed scVI, trVAE and ACTINN, while running efficiently on standard GPUs rather than requiring HPC infrastructure.
4️⃣ One foundation, many tasks
Annotation, harmonisation, embedding and imputation are all handled by the same model, removing the need for repeated task-specific training runs.
💡 Why It’s Cool
scPRINT2 moves the field closer to something single-cell biology has been missing: a shared base model that is practical, reusable and trained on the kinds of data researchers actually work with. It is not perfect, but it is a meaningful step towards standardised foundations rather than endless retraining.
📄 Read the paper
⚙️ Explore the model
🧪 DynaMate: Autonomous Molecular Dynamics Without Human Supervision
What if an AI agent could plan, run and analyse protein–ligand MD simulations on its own?
Force field-based molecular dynamics is essential for understanding protein–ligand behaviour, but setting up simulations remains slow and error-prone. Structure preparation, parameterisation, software quirks and debugging often turn MD into a full-time job.
DynaMate, developed by the team at EPFL, is a modular multi-agent system that automates the entire MD workflow. It plans simulations, executes them and analyses results for protein-only and protein–ligand systems, without human input.
Think of it as a simulation workflow with a built-in research assistant.
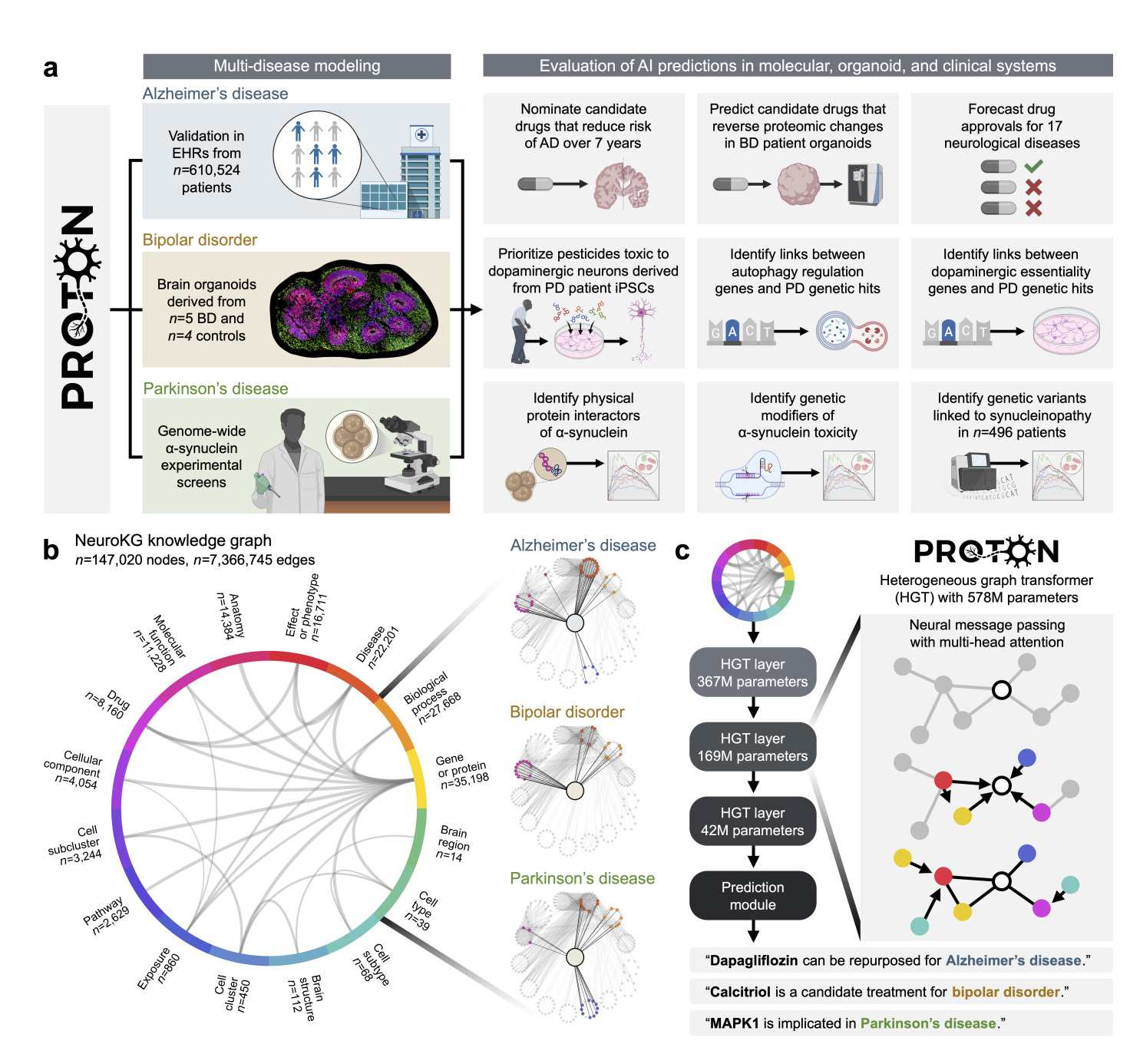
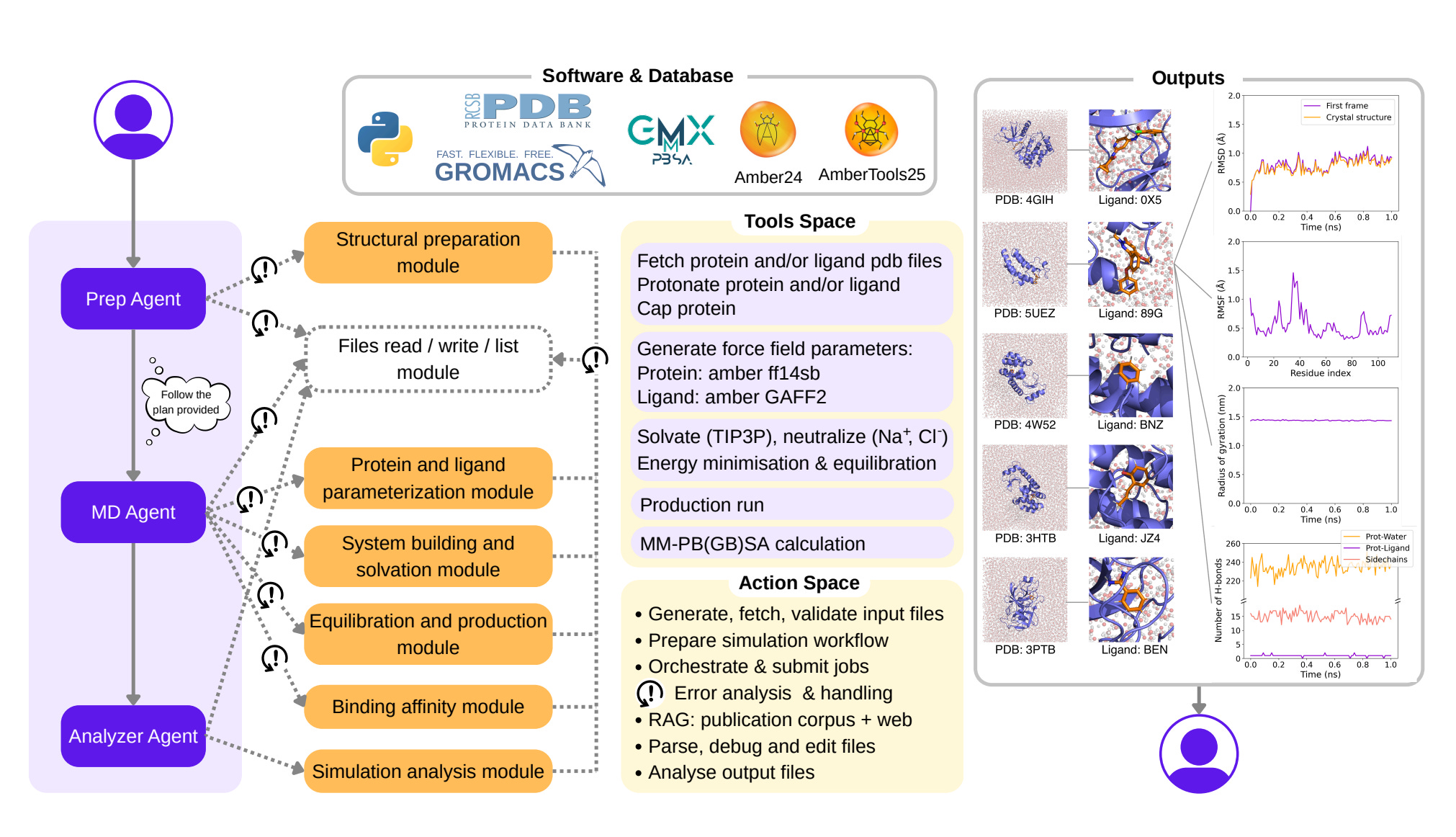
🔬 Applications and Insights
1️⃣ Autonomous MD workflows for complex systems
DynaMate successfully ran 12 full MD simulations, including protein–ligand complexes such as BRD4 and trypsin. Ten of the twelve systems completed with a 100 per cent success rate and no user prompts.
2️⃣ Built-in error detection and correction
When simulations failed, agents adapted rather than stopping. One agent rewrote force field files to fix atom naming issues, while another removed unnecessary restraints to stabilise runs.
3️⃣ Binding affinity predictions that rival docking
Across ten BRD4 inhibitors, DynaMate’s MM PBSA binding energies correlated with experimental IC50s at r = 0.597, outperforming GNINA’s r = 0.385.
4️⃣ Specialised agents for setup, execution and analysis
The system splits tasks across a planner, an MD runner and an analyser, each using tool reasoning, literature search and software integration with GROMACS and AmberTools.
💡 Why It’s Cool
DynaMate is not just automation. It is automation that reasons. The system detects failures, fixes them and explains what went wrong. Even when it fails, it fails productively. That is a meaningful shift for computational chemistry workflows.
📄 Read the preprint
⚙️ Try the code
⚛️ PROTON: Teaching Models to Explain Why Molecules Bind
What if a model could explain why a molecule binds, not just score it?
Generative models can propose molecules at scale, but most struggle to explain binding mechanisms or guide rational optimisation. They predict outcomes without offering insight.
PROTON, developed by researchers at Harvard, takes a different approach. It is a language model trained to reason about protein–ligand interactions, combining sequence, structure and chemistry into a unified representation while providing interpretable explanations.
🔬 Applications and Insights
1️⃣ Joint reasoning over sequence, structure and chemistry
PROTON ingests protein sequences, 3D binding pockets and ligand graphs, fusing them into a shared embedding space to predict binding and highlight interaction hotspots.
2️⃣ Strong benchmark performance
On the LIT-PCBA benchmark, PROTON achieves an AUROC of 0.92 and outperforms structure-only models such as PocketGCN and BANANA across multiple targets.
3️⃣ Large-scale self-supervised pretraining
The model is pretrained on 180,000 proteins using masked language modelling and contrastive learning, allowing it to learn protein–ligand interaction patterns even without labelled binding data.
4️⃣ Interpretable binding site attribution
Attention scores highlight key binding residues, aligning closely with crystallographic evidence across test cases.
💡 Why It’s Cool
We do not just need more molecules. We need models that understand why they work. PROTON treats molecular interaction as a language problem, opening the door to explanation-driven design rather than blind optimisation.
📄 Read the preprint
⚙️ Explore the code
Thanks for reading Kiin Bio Weekly!
💬 Get involved
We’re always looking to grow our community. If you’d like to get involved, contribute ideas or share something you’re building, fill out this form or reach out to me directly.
Connect With Us
Have any questions or suggestions for a post? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website