
MIT's SwitchCraft, Shanghai Jiao Tong's TadA-Bench, and Helmholtz Munich's Chem-PerturBridge
Kiin Bio's Weekly Insights

Welcome back to your weekly dose of AI news for Life Science!
Three papers this week that all ask some version of the same question: are we actually measuring the right thing? SwitchCraft pushes protein design past the single-structure assumption that underpins most current methods. TadA-Bench asks whether protein language models can predict the future of evolution or only interpolate its past. Chem-PerturBridge harmonises the messy pile of perturbation transcriptomics data and reveals just how little of it agrees at the gene level. The connecting thread: the foundation models exist, but the design paradigms and benchmarks haven’t caught up.
We just opened up our Kiin Pioneer Programme: free access to our platform for academic and nonprofit research teams for a year.
The short version: we’ve built a place where scientists can collaborate on their drug discovery work without everything living in disconnected tools and someone’s local files. Literature reviews, target discovery, bioinformatics, all in one place. When one person finds something interesting and someone else has relevant data, the platform catches that and suggests what to look at next. Everything’s tracked, so six months later you actually know what was done and why.
We’re looking for teams who are trying to move faster on questions like: which targets should we prioritise? How do we make sense of conflicting evidence? What’s actually worth testing next?
No cost, no data transfer, all IP stays with your institution. Applications close in August, first cohort starts in September.

SwitchCraft: A Programmatic Framework for Designing State-Switching Proteins
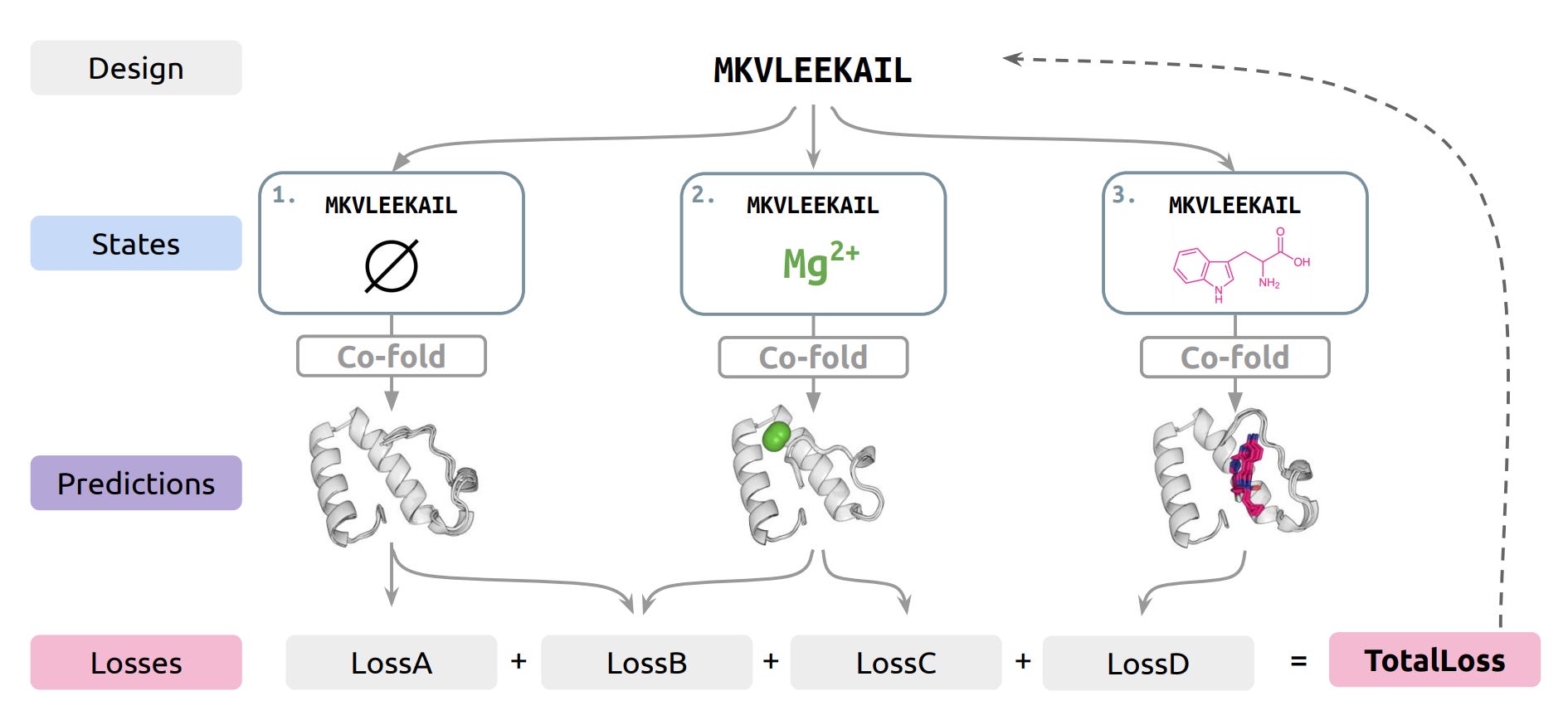
🔬 Protein design tools optimise for one structure. Real proteins switch between conformations to do their jobs: allosteric enzymes toggle between active and inactive states, biosensors change shape upon binding. Designing proteins that intentionally switch between defined states has been a manual, low-throughput exercise.
Jing, Bafna, and colleagues from MIT’s Berger lab built SwitchCraft, a framework that designs proteins with specified multi-state behaviour by backpropagating through differentiable structure prediction models.
🧬 The core idea: treat Boltz-1 (an open-source structure prediction model) as a differentiable loss function. Define the desired structural states, then optimise the sequence so that it folds into all of them under appropriate conditions. The framework is programmatic. You compose design objectives from modular building blocks rather than training a new model per task.
⚡ They demonstrate allosteric regulation of protein motifs, discrimination between bound ligand identities, and fluorescent biosensor design. The biosensor results are particularly telling: designed sequences show distinct fluorescence states depending on which ligand is bound. That’s functional multi-state behaviour that hasn’t been accessible through standard single-state design pipelines.
🧪 Where This Fits
This sits at the leading edge of computational protein design, downstream of structure prediction but upstream of experimental characterisation. The reason it exists now is simple: differentiable structure prediction. Until models like Boltz-1, ESMFold, and AlphaFold became fast and accurate enough to use as gradient-providing modules, you couldn’t backpropagate through structure. RFdiffusion and ProteinMPNN design single structures beautifully, but they don’t handle the multi-state problem. SwitchCraft does something conceptually different: it treats structure prediction as a subroutine rather than the end goal. The limitation is that experimental validation here is still computational, relying on Boltz-1’s predictions of the designed states. Whether these sequences actually fold into multiple states in a wet lab remains open, though the biosensor designs are testable. If you work on biosensors, allosteric switches, or molecular logic gates, this is worth trying now.
💡 Why This Is Cool
This is what it looks like when structure prediction becomes infrastructure rather than the main event. The field spent five years building accurate folding models. Now those models are components in design loops. Multi-state protein design has been a goal since the Kuhlman lab’s early work on conformational switches, but the computational tools never matched the ambition. SwitchCraft doesn’t solve the full problem (experimental validation is still the bottleneck), but it makes the design step tractable in a way it simply wasn’t before.
📄 Read the paper.
💻 Try the code.
TadA-Bench: A Million-Variant Benchmark for Future-Round Discovery Toward Agentic Protein Engineering
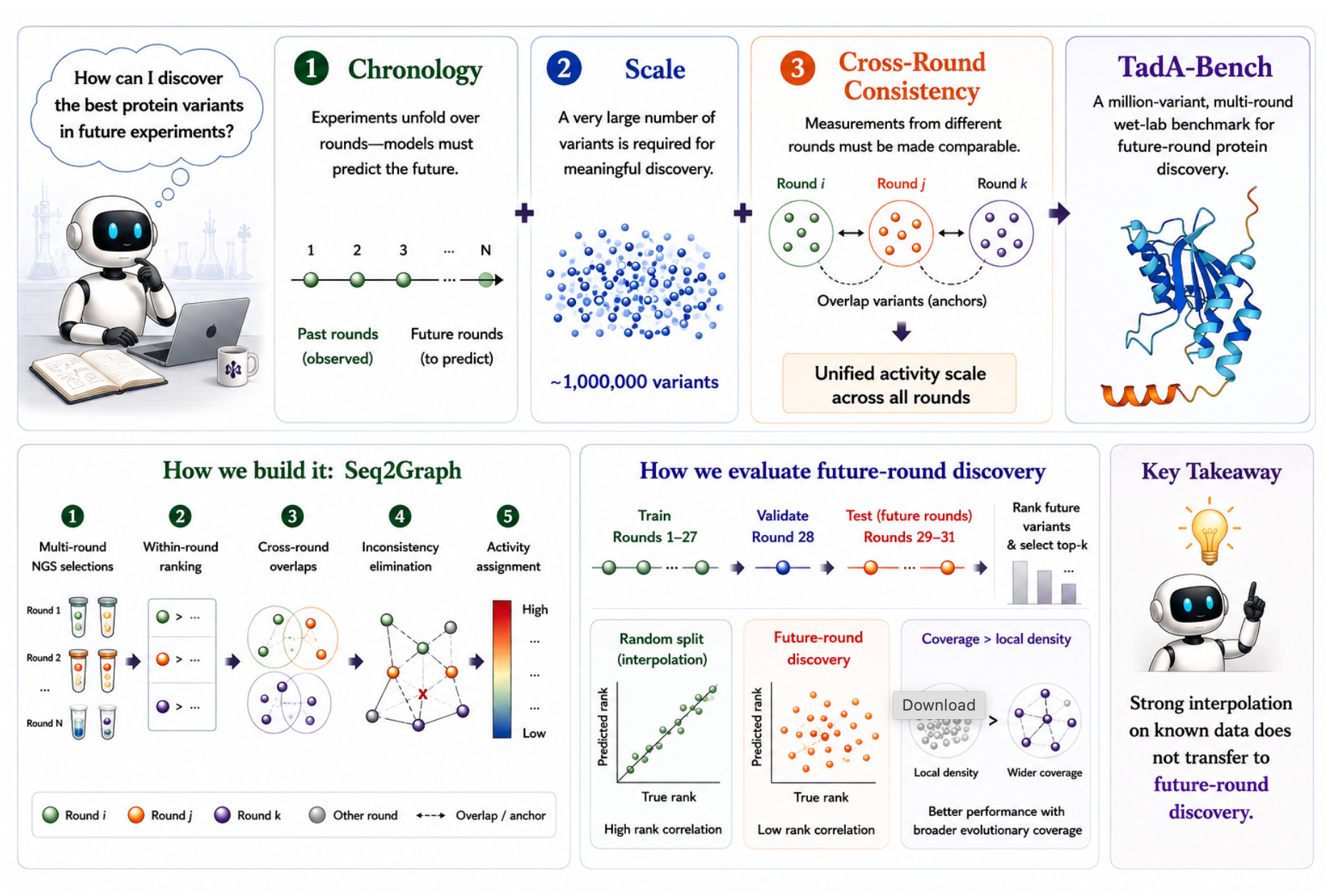
🔬 Protein fitness prediction benchmarks typically test whether models can fill in gaps within a mutational landscape. That’s interpolation. What protein engineers actually need is extrapolation: given rounds 1 through 10 of directed evolution, can you predict what we’ll find useful in round 11? No existing benchmark properly tests this.
Gao and colleagues from Shanghai Jiao Tong University built TadA-Bench from 31 rounds of real TadA (tRNA adenosine deaminase) directed evolution, totalling roughly one million variants.
🧬 The benchmark enforces chronological evaluation: models train on earlier rounds and must predict which variants from later rounds will be experimentally validated. It provides aligned DNA, RNA, and protein sequences, and uses a Seq2Graph method to create comparable activity measurements across rounds that originally used different assay conditions.
⚡ The headline finding is sobering. Current protein language models (including ESM-2 and various fine-tuned variants) struggle with temporal prediction even when they perform well on standard interpolation benchmarks. The gap between interpolation and extrapolation performance is substantial. Good performance on DMS datasets doesn’t mean your model can guide the next round of experiments. Many practitioners suspected this; now there’s a number attached to it.
🧪 Where This Fits
This is a benchmark, not a tool, and its value depends on adoption. It fills a gap the field has known about for years. ProteinGym and similar resources test interpolation well, but nobody had assembled a large-scale temporal benchmark from real directed evolution campaigns. The 31 rounds of TadA evolution provide unusually rich temporal structure (most published datasets have 3-5 rounds at most). The “agentic protein engineering” framing is timely: as more groups wire LLMs into experimental design loops, you need evaluation frameworks that test whether the model’s suggestions actually lead somewhere productive. The dataset is on Hugging Face and the code is open on GitHub, which lowers the barrier to adoption. The main caveat is generalisability: TadA is one enzyme. Performance on this benchmark won’t guarantee performance on your protein of interest. Still, it’s the best temporal test we have.
💡 Why This Is Cool
The protein ML field has a measurement problem. Papers report NDCG on held-out DMS positions and claim their model “guides protein engineering.” This benchmark calls that bluff. Can your model predict the future, or only reconstruct the past? The distinction matters enormously for anyone running a real directed evolution campaign. If current models fail at temporal prediction (and they largely do), that’s uncomfortable but useful information. It tells you where the actual research gap is, which is more valuable than another leaderboard.
📄 Read the paper.
🤗 Access the dataset.
Chem-PerturBridge: A Harmonized Compendium of Small Molecule Perturbation Transcriptomic Effects
🔬 The field has generated enormous amounts of small molecule perturbation transcriptomics data (L1000, sci-Plex, Tahoe, and many smaller datasets). The problem: nobody knows how well they agree with each other, and combining them for model training requires harmonisation that hasn’t been done systematically.
Szałata and colleagues from the Theis lab at Helmholtz Munich built Chem-PerturBridge, standardising 37,000+ compounds across 1.25 million samples from eight assay types with consistent metadata.
🧬 The compendium spans nine datasets including sci-Plex3, Tahoe, L1000, OP3, DILImap, and others. The harmonisation pipeline normalises cell line annotations, compound identifiers, and gene nomenclature, making cross-dataset comparison possible for the first time at this scale.
⚡ The uncomfortable finding: gene-level agreement between datasets is poor. When the same compound is tested in the same cell line across different platforms, correlation at the individual gene level is low. Directional consistency (is the gene up or down?) is better, and turns out to be sufficient for improving compound representation learning. Models trained on the harmonised directional data outperform those trained on any single dataset alone

🧪 Where This Fits
This sits upstream of almost everything in computational chemical biology: virtual screening, target identification, mechanism-of-action prediction, and toxicity forecasting all depend on perturbation data quality. The honest finding here is that the perturbation transcriptomics field has a reproducibility problem at the gene level, even between high-quality experiments. That’s not new as a suspicion, but quantifying it across eight assay types is valuable. The positive takeaway is that directional signals persist, and they’re enough to learn useful compound representations. For practitioners, this means you should probably stop training on raw gene-level expression values from a single perturbation study and start using directional features from multiple sources. The resource is open (MIT licence on code, upstream licences on data), which is appropriate for something positioned as community infrastructure.
💡 Why This Is Cool
The field has been treating perturbation transcriptomics datasets as interchangeable training data without checking whether they actually say the same thing. They don’t, at least not at the resolution most people assume. This paper does the unglamorous work of quantifying that disagreement and showing what signal does survive. The practical consequence: if you’re building models on LINCS L1000 data alone, you’re probably leaving performance on the table. Directional agreement across platforms is a more robust training signal than absolute expression values from one platform. This is infrastructure work rather than a methods advance, but it’s the kind that makes everything downstream more trustworthy.
📄 Read the paper.
💻Try the code.
🗓️ Events & Competitions
The best competitions, hackathons, and community challenges in AI x life sciences, curated weekly. Know something worth featuring? Reply and let us know.
More upcoming events:
Creative Disruption Forum: Modern Drug Discovery | June 18, NIAB Cambridge
A full-day forum for biotech and R&D leaders exploring how technology is changing small molecule drug discovery. Keynote interviews with industry thought leaders followed by workshops under Chatham House Rules, limited to 60 attendees. Part of Cambridge Wide Open Week. Organised by Graham Combe and Prof Tony Sedgwick. £60 for biotech companies.
London Protein Design Day | June 23, Imperial College London
The first edition of a one-day symposium bringing together London’s protein design community and beyond. Programme spans AI-driven design, molecular dynamics, and bioinformatics, with applications across enzymes, antibodies, and materials. Organised by Pietro Sormanni, Rebecca Birolo, and Jakub Lála. Abstract deadline for poster/oral presentations is this Saturday (May 17). In person only.
BioHackathon Europe 2026 | November 9-13, Barcelona
ELIXIR’s annual international bioinformatics hackathon, running since 2018. 160+ participants, five days of collaborative coding on open bioinformatics infrastructure and tools. The call for project proposals opens March 16 and closes April 15 - so if you want to lead a project, that’s your window.
Thanks for reading!
💬 Get involved
We’re always looking to grow our community. If you’d like to get involved, contribute ideas or share something you’re building, fill out this form or reach out to me directly.
Connect With Us
Have questions or suggestions? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website