
Modena's G4REP, Harvard's evedesign, and Purdue's Peptide-Protein Docking Review
Kiin Bio's Weekly Insights

Welcome back to your weekly dose of AI news for Life Science!
Keeping up with AI x life science news can get exhausting.
It’s scattered across LinkedIn, X, Substack, arXiv, Slack, newsletters... and you still somehow miss the things that actually matter. Too much noise, not enough signal.
We’re building something to fix that: a smarter, more powerful way to stay on top of what’s actually relevant to you.
But we want to build it with you, not just for you. Take 2 minutes to tell us what’s missing. What you share will directly shape what we build, and you’ll be the first to benefit from it.
G4REP: RNA G-quadruplex-binding protein prediction across the human proteome
🧬 RNA G-quadruplex-binding proteins regulate mRNA processing, localisation, and stress responses - but experimental detection alone can’t scale. G4REP maps them across the entire human proteome.
🔬 RNA G-quadruplexes act as post-transcriptional regulatory hubs, but identifying which proteins bind them is experimentally challenging. Classical approaches rely on known binding domains like zinc fingers, missing a wide range of RG4-interacting proteins.
Researchers at the Università degli Studi di Modena e Reggio Emilia and Sapienza Università di Roma introduce G4REP, combining ESM-2 embeddings with LSTM neural networks to predict RG4-binding proteins at proteome scale.
🧬 G4REP analyses protein sequences for RG4 binding features: arginine-glycine-rich motifs, intrinsically disordered regions, and aromatic residues, identifying the short flexible motifs through which interactions typically occur.
⚡ ~85% accuracy and AUROC of 0.91. Over 2000 candidate RG4-binding proteins identified across the human proteome, including 552 high-confidence hits.

🔬Applications and Insights
1️⃣ Identifying Therapeutic Targets
RG4-binding proteins are enriched in stress granules linked to cancer and neurodegeneration. G4REP identifies candidates with ~85% accuracy, enabling reliable prioritisation of targets influencing RNA stability and translation.
2️⃣ Expanding RNA-Binding Protein Networks
G4REP identified 2000+ candidate proteins including poorly characterised FAM families, expanding RNA regulation well beyond classical binding domains.
3️⃣ Predicting Functional Binding Regions
G4REP pinpoints binding sites within proteins using a disorder-weighted residue score, highlighting short RGG-rich motifs as primary interaction sites.
4️⃣ Understanding Cellular Stress Responses
552 high-confidence RG4-binding proteins localise to stress granules, supporting a coordinating role for RG4 interactions in RNA metabolism during cellular stress.
💡 Why This Is Cool
G4REP opens up RG4 biology at a scale experimental methods alone cannot reach. It expands our map of RNA-protein interactions and surfaces previously hidden regulators and therapeutic candidates across the human proteome.
📖 Read the paper
💻 Code: https://lnkd.in/gYTkHmRd
evedesign: accessible biosequence design with a unified framework
🧬 Protein design has dozens of ML models. None of them talk to each other. Every real-world project still requires custom glue code, reformatting, and one-off pipelines. evedesign changes that.
🔬 The design problems that matter most in protein engineering - conditional design under real-world constraints, multi-objective optimisation, and iterative lab-in-the-loop workflows - demand flexible, composable infrastructure that no single tool provides. Current ML methods are rarely interoperable and remain inaccessible to non-experts.
Researchers at Harvard Medical School, Wellcome Sanger Institute, University of Cambridge, Seoul National University, Broad Institute, and collaborators built evedesign, a unified open-source framework that formalises conditional biosequence design in a method-agnostic way.
🧬 evedesign defines three composable operations - Generate, Score, and Transform - that work across any model type (MSA-based, LLM, inverse folding, de novo 3D). A standardised multi-level instance representation (sequence, embedding, and 3D structure simultaneously) lets outputs from one model feed directly into another without reformatting.
⚡ Supports multi-objective optimisation, supervised and unsupervised model integration, and lab-in-the-loop iteration from the ground up. Interactive web interface at evedesign.bio takes users from target protein to orderable DNA. Demonstrated in antibody engineering, enzyme design, and natural enzyme discovery. MIT-licensed, FAIR-compliant, and self-hostable.
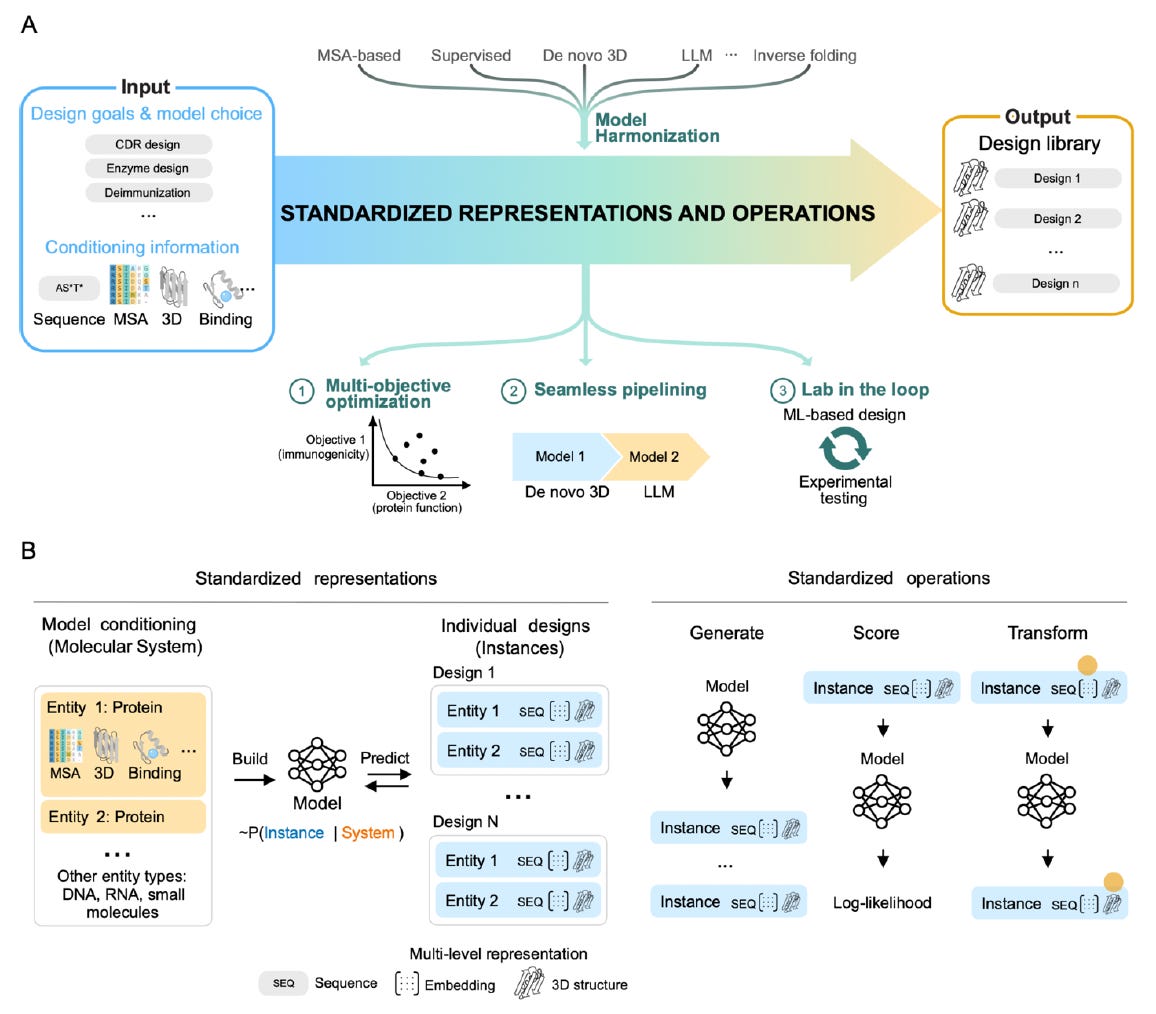
🔬Applications & Insights
1️⃣ Antibody Engineering Conditional
CDR design subject to multiple constraints (thermostability, deimmunisation, binding) using composable multi-objective pipelines - no custom code required.
2️⃣ Enzyme Design
Combine generative models with scoring functions from different method families in a single workflow, iterating with experimental data as it comes in.
3️⃣ Lab-in-the-Loop Workflows
Declarative, serialisable pipelines that can halt, incorporate new experimental results, and resume - built for iterative design rounds rather than one-shot prediction.
4️⃣ Accessibility for Non-Computational
Researchers End-to-end web interface makes ML-driven protein design accessible to experimentalists without requiring programming or model-specific expertise.
💡 Why This Is Cool
The bottleneck in protein engineering isn’t any individual model - it’s connecting them. evedesign is the first open-source framework that treats this as a first-class problem: standardised interfaces, composable workflows, and lab-in-the-loop iteration built in from the start. That’s infrastructure the field has needed for years.
📖 Read the paper
💻 Website
Peptide-protein docking: from physics-based models to generative intelligence
🔬 Peptide therapeutics are one of the fastest-growing drug modalities - they target flat, extended protein surfaces that small molecules can’t reach. But computationally predicting how a peptide binds its target remains hard: peptides are flexible, often disordered, and fold upon binding. Classical docking methods struggle with all three.
Researchers at Purdue University and Korea University College of Medicine review the full landscape of peptide-protein docking, from traditional physics-based approaches to the latest deep learning methods, covering 25+ tools across three generations.
🧬 The review organises methods into three categories: binding site predictors that guide or filter docking models, AlphaFold-based protocols for peptide-protein co-folding and refinement, and deep generative models (diffusion-based) that sample peptide conformations conditioned on a target structure.
⚡ Diffusion models like RAPiDock and DiffPepDock are emerging as the most promising direction, handling peptide flexibility natively. AlphaFold-based methods work well for structured peptides but struggle with disordered ones. Major remaining gaps: limited training data, weak performance on long peptides, and almost no methods handle chemically modified peptides.
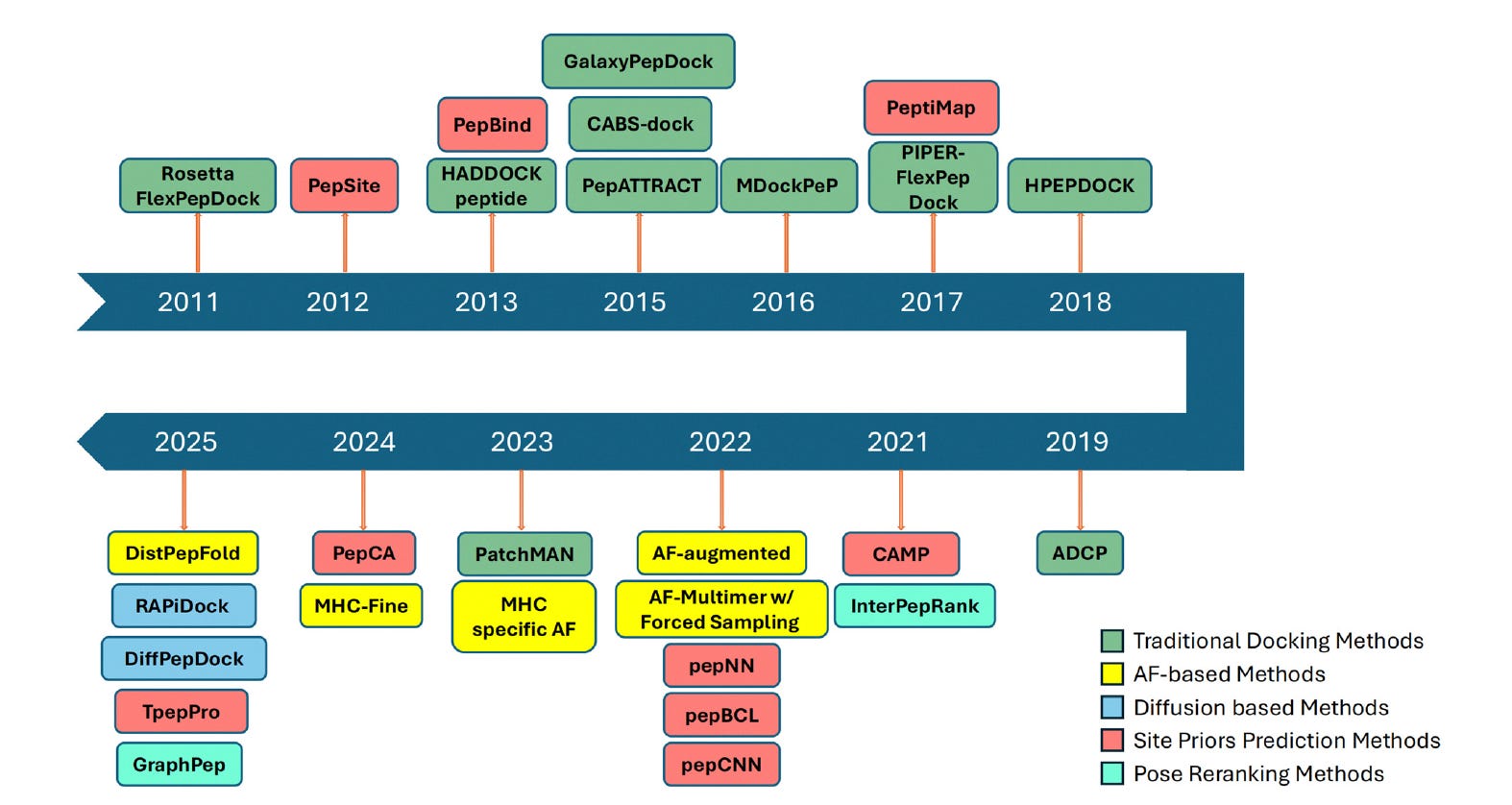
🔬Applications & Insights
1️⃣ Peptide Drug Design
Maps the full computational toolkit for predicting peptide-protein binding modes - essential for designing peptide therapeutics against previously undruggable protein surfaces.
2️⃣ Masking Peptide Engineering
Covers tools applicable to masking peptides that block drug binding sites until protease activation at the tumour - a growing immunotherapy strategy.
3️⃣ Practical Method Selection
Table 1 catalogues 25+ methods with years, descriptions, and code availability links - a ready-made decision guide for choosing the right docking approach.
4️⃣ Open Challenges
Identifies the three biggest gaps: training data scarcity, long/disordered peptide performance, and chemical modification handling - a roadmap for future method development.
💡 Why This Is Cool
This is the review the peptide docking field needed. It doesn’t just catalogue methods - it explains when each approach works, when it breaks, and what’s missing. If you’re working on peptide therapeutics, this is your decision guide for computational docking in 2026.
📖 Read the paper
🗓️ Events & Competitions
The best competitions, hackathons, and community challenges in AI x life sciences, curated weekly. Know something worth featuring? Reply and let us know.
🏗️ Hackathon Highlight
The Elnora x Monomer Bio AI scientist hackathon brought together 43 builders for 24 hours of agent-driven lab automation. The winning team connected Elnora’s protocol generation agent to a robotic arm that autonomously imaged cells, assessed confluency at 70%, generated a splitting protocol, executed it, and re-imaged to confirm results - a fully closed AI-to-robot lab loop built from scratch overnight. 6 out of 7 teams incorporated Elnora into their workflows.
Full recap from CEO Carmen Kivisild.
Recap on Edinburgh BioHackathon
Last weekend, Edinburgh hosted its first ever biohackathon, and the numbers speak for themselves.
🎯 350 applications. 110 selected participants. Over 95% attendance rate, which for a free hackathon is exceptional (typical rates sit around 40-50%).
Organised entirely from scratch by a volunteer team led by Xi Yang AMRSC and Yen Peng Chew, PhD, AFHEA at the The University of Edinburgh, BioHackathon Edinburgh (PRIMED) brought together students, postdocs and clinical researchers from across Scotland: 52% from The University of Edinburgh, 48% from University of Dundee, University of St Andrews, University of Stirling, University of Glasgow and beyond.
Seven challenges spanned three tracks: academic, industry and non-coder. The two standouts were the bio-business and Pacifico Biolabs (Genome-Scale Metabolic Modelling Tool) track, which saw the highest participation with 8 and 7 teams respectively.
🔬 One project that caught everyone's attention: FilamentTracker. A team built a fully functional web tool with its own custom domain that detects and tracks protein filaments in microscopy images, showing filament length and area over time. In 48 hours. Judges flagged it as potentially publishable if stress-tested across other organisms.
What made this event different was the deliberate push for interdisciplinary collaboration. Teams were encouraged to mix engineers, biologists, data scientists and non-coders. Not everyone landed in a mixed team, but the feedback was clear: those who did got the most out of it.
All submissions are open source on DevPost (https://lnkd.in/gjpQwXXR). Next up from the organisers: peer-led company creation workshops to help winning teams take their projects further.
🚀 Scotland's first biohackathon, built from a blank Google Drive folder by an unpaid volunteer team. If this is what version one looks like, version two is going to be something special!
More upcoming events:
BioHackathon Europe 2026 | November 9-13, Barcelona
ELIXIR’s annual international bioinformatics hackathon, running since 2018. 160+ participants, five days of collaborative coding on open bioinformatics infrastructure and tools. The call for project proposals opens March 16 and closes April 15 - so if you want to lead a project, that’s your window.
Thanks for reading!
💬 Get involved
We’re always looking to grow our community. If you’d like to get involved, contribute ideas or share something you’re building, fill out this form or reach out to me directly.
Connect With Us
Have questions or suggestions? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website