
Oxford's MolJSON, DTU's PlaTITO, and OpenBind's Dataset Release
Kiin Bio's Weekly Insights

Welcome back to your weekly dose of AI news for Life Science!
Keeping up with AI x life science news can get exhausting.
It’s scattered across LinkedIn, X, Substack, arXiv, Slack, newsletters... and you still somehow miss the things that actually matter. Too much noise, not enough signal.
We’re building something to fix that: a smarter, more powerful way to stay on top of what’s actually relevant to you.
But we want to build it with you, not just for you. Take 2 minutes to tell us what’s missing. What you share will directly shape what we build, and you’ll be the first to benefit from it.
🇺🇸 We’re heading to Bio-IT World in Boston, May 19-21.
Our CEO Filippo and CTO Bogdan will be there and would love to meet anyone thinking about:
How AI is actually changing preclinical workflows (not just the hype)
Why drug discovery is a systems problem, not just a science one
What it takes to go from 5-year timelines to something radically faster
No pitch, just good conversation. If any of that’s on your mind, reach out - we’ll find a time to grab a coffee.
MolJSON: Molecular Representations for Large Language Models
🔬 LLMs are increasingly used in chemistry for tasks like reaction prediction and structure elucidation, but they need to read and write molecular structures reliably. Previous work has defaulted to SMILES strings or IUPAC names, but no one has systematically tested whether these formats are actually good for LLMs. Both impose strict serialisation rules that may not align with how language models process information.
Researchers at Oxford introduce MolJSON, a structured JSON schema that represents molecular graphs explicitly as lists of atoms and bonds. Unlike SMILES (which requires a specific graph traversal) or IUPAC (which requires rule-based nomenclature), MolJSON presents the molecular graph directly in a format compatible with LLM structured output modes.
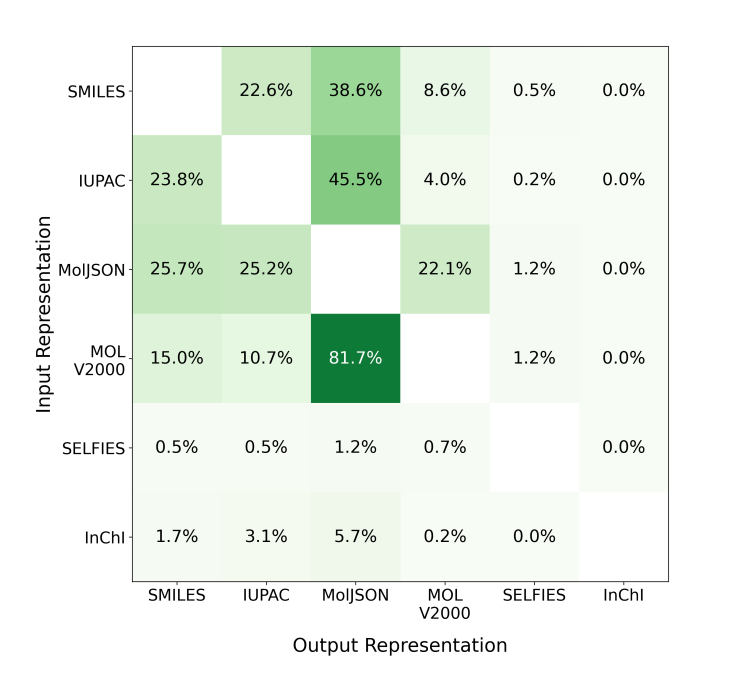
🧬 They evaluated five molecular representations across 78,045 algorithmically generated questions spanning translation, shortest-path reasoning, and constrained generation tasks using GPT-5-nano, GPT-5-mini, GPT-5, and Claude Haiku 4.5. MolJSON consistently outperformed all existing formats as both an input and output representation.
⚡ On translation tasks, GPT-5 achieved 71.0% accuracy converting IUPAC to MolJSON versus 43.7% for IUPAC to SMILES. For constrained generation, GPT-5 reached 95.3% accuracy generating MolJSON versus 64.0% for SMILES and 76.3% for IUPAC. MolJSON was also 1.8x more token-efficient than SMILES on reasoning tasks. Performance advantages held even though models were never explicitly trained on MolJSON.
🔬 Applications and Insights
1️⃣ Better Chemistry Agents
LLM-based chemistry systems that read and write molecules can operate more reliably by switching to MolJSON, reducing errors from format parsing failures.
2️⃣ Robust Molecular Reasoning
MolJSON maintained high accuracy even on complex molecules with fused rings and high atom counts, where SMILES and IUPAC performance degraded sharply.
3️⃣ Token-Efficient Representations
Graph-based formats let models skip the internal reconstruction step needed for traversal-based representations, using fewer reasoning tokens and reducing latency.
4️⃣ Format-Agnostic Improvement
MolJSON outperformed SMILES and IUPAC despite those formats being well-represented in LLM training data, suggesting explicit graph encodings are intrinsically better aligned with how LLMs process structure.
💡 Why This Is Cool
Everyone building LLM chemistry tools has been using SMILES because it was already there. This paper shows that is leaving significant performance on the table. A simple change in molecular representation, with no model retraining, unlocks dramatically better accuracy across translation, reasoning, and generation. The fact that LLMs spontaneously generate semantically meaningful atom identifiers (like “C_acyl” or “Npip”) in MolJSON suggests these models can reason about molecular graphs more naturally when given an explicit graph format.
📄 Read the paper
💻 Try the code
PLaTITO: Protein Language Model Embeddings Improve Generalisation of Implicit Transfer Operators
🔬 Molecular dynamics simulations are essential for understanding protein behaviour, but conventional MD is computationally prohibitive for biologically relevant timescales. Generative molecular dynamics methods learn surrogate models from trajectory data, but they typically require large collections of long MD trajectories and struggle to generalise to unseen protein systems.
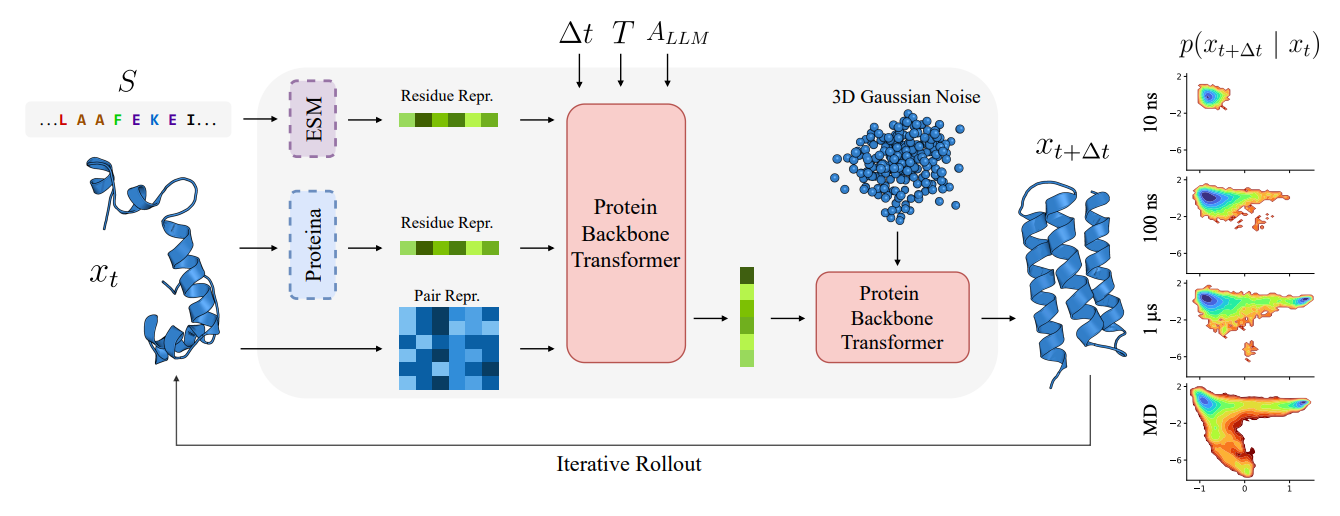
PLaTITO from Chalmers, Copenhagen, and DTU introduces coarse-grained transferable implicit transfer operators (TITO) for protein molecular dynamics that generalise to out-of-distribution protein systems. By conditioning on protein language model embeddings from ESM and structure embeddings from Proteina, the model learns to transfer across diverse proteins without system-specific fine-tuning.
🧬 Trained on the mdCATH dataset (4,471 domains, ~56 ms aggregate simulation time), PLaTITO learns long-time transition densities conditioned on backbone coordinates, sequence, temperature, and time step. The architecture uses a two-stage design: a conditioning network produces per-residue representations, and a velocity network generates the flow field for sampling future conformations.
⚡ PLaTITO-Big (19M parameters) outperforms BioEmu across all equilibrium sampling metrics on fast-folding proteins while requiring substantially less training data (56 ms vs. 216 ms) and compute (1,100 GPU hours vs. 9,216). It recovers non-Arrhenius temperature-dependent folding kinetics and explores cryptic binding pockets, generating trajectories with repeated folding and unfolding events at microsecond timescales.
🔬 Applications and Insights
1️⃣ Out-of-Distribution Generalisation PLaTITO transfers to unseen proteins without fine-tuning, unlike Boltzmann Emulators that require system-specific training data.
2️⃣ Data-Efficient Training Achieving state-of-the-art equilibrium sampling with 4x less MD data and 8x less compute than BioEmu demonstrates that transfer learning can dramatically reduce the data barrier.
3️⃣ Temperature-Dependent Kinetics Explicitly conditioning on temperature lets PLaTITO capture non-Arrhenius behaviour, reproducing physically meaningful folding and unfolding rates across temperature ranges.
4️⃣ Cryptic Binding Pocket Discovery PLaTITO-Big samples conformational transitions to cryptic pockets from both apo and holo states, opening applications in drug discovery for targeting hidden binding sites.
💡 Why This Is Cool
This is the first transferable molecular dynamics model that genuinely generalises across protein systems while beating dedicated Boltzmann Emulators on their own benchmarks. The key insight is that pretrained protein language models encode enough structural and evolutionary information to let a small (3-19M parameter) dynamics model transfer across diverse folds. Generating realistic microsecond-scale folding trajectories on a single GPU in seconds, rather than months of conventional simulation, changes what is computationally accessible for studying protein dynamics.
📄 Read the paper
OpenBind: A Structure-Affinity Dataset for Structure-Based AI in Drug Discovery
🔬 Structure-based AI for drug discovery is held back by a data bottleneck. Public protein-ligand datasets are sparse, unevenly distributed, and rarely link crystallographic binding modes with quantitative affinity measurements at scale. Current ML methods for docking, cofolding, and affinity prediction are difficult to evaluate fairly because most benchmarks overlap with training data.
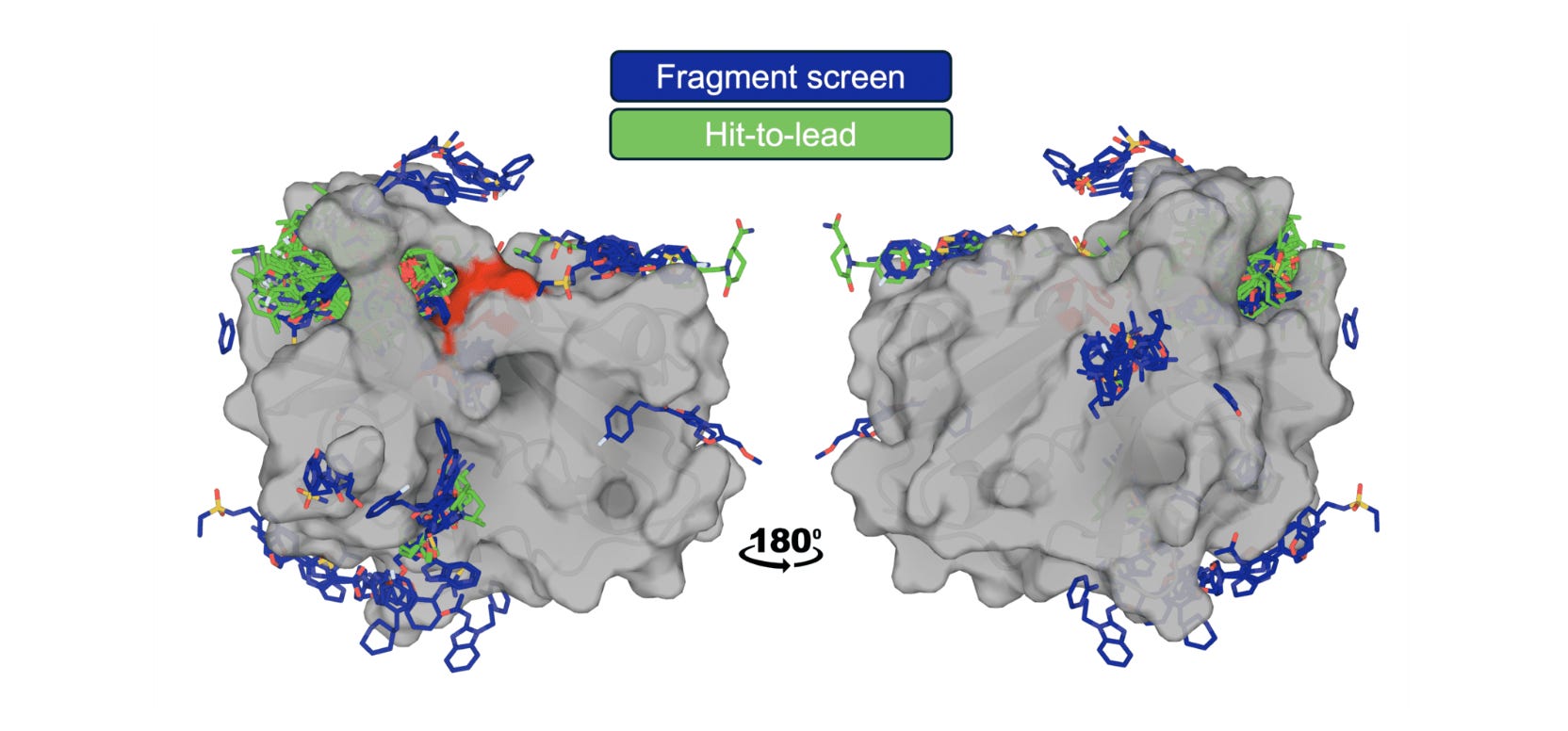
The OpenBind consortium (Diamond Light Source, Oxford, and partners) releases a dense structure-affinity dataset: 925 crystallographic binding events from 699 compounds with affinity measurements for 601 compounds, all targeting EV-A71 2A protease, a viral target relevant to pandemic preparedness. The data are deliberately positioned in an under-represented region of protein-ligand space relative to existing public structures.
🧬 The dataset includes fragment screen hits and follow-on compounds with KD values from Creoptix WAVEsystem measurements, creating a coherent experimental series where users can study local structure-activity relationships. Reference benchmarks span conventional docking (AutoDock Vina), ML docking (GNINA, DiffDock), cofolding (AlphaFold3, Boltz, OpenFold3), and affinity prediction methods.
⚡ Redocking achieves up to 85% success (GNINA), but cross-docking into apo structures drops below 5% due to binding-site loop conformational changes. Cofolding methods reach 36% (OpenFold3-p2), but fine-tuning on fragment-screen data boosts this to 76%, approaching redocking performance. For affinity prediction, a simple molecular-weight baseline outperforms most structure-based methods, highlighting how challenging this task remains.
🔬 Applications and Insights
1️⃣ Benchmarking Beyond Training Data
The EV-A71 2A protease complexes are dissimilar to pre-2021 PDB data, providing a genuine test of whether cofolding and docking methods generalise or just memorise near-neighbours.
2️⃣ Fragment Screens as Training Data
Fine-tuning cofolding models on fragment-bound structures doubled success rates on follow-on compounds, showing that early experimental data can feed directly into AI model improvement.
3️⃣ Separating Failure Modes
The dataset cleanly distinguishes receptor-conformation failures from ligand-placement failures, letting method developers target specific weaknesses rather than debugging aggregate metrics.
4️⃣ Affinity Prediction Reality
Check Simple baselines beating structure-based methods on this dataset is a clear signal that current affinity models may be learning chemical trends rather than genuine protein-ligand interaction physics.
💡 Why This Is Cool
OpenBind is not just releasing more structures. It is building the experimental infrastructure to generate the kind of data that structure-based AI actually needs: dense, linked structure-affinity measurements within coherent chemical series, positioned where current models are weakest. The fragment fine-tuning result is particularly striking. It shows that a relatively small crystallographic screen can transform cofolding performance on a new target, pointing toward a practical workflow where early experiments directly improve computational predictions for the same campaign.
📄 Read the data
💻 Try the benchmarks
📬 Newsletter Shout-Out
This week we're shouting out Building in BioAI, a monthly newsletter from Joe:
Building in BioAI is a monthly newsletter for those operating in, or interested in, the AI-enabled biology space. That’s founders, technical leaders, and individual contributors working within areas like therapeutics, diagnostics, and tooling.
Joe’s roundup centres on observations from within the space, including analysis of how teams are structuring themselves, what’s changing in hiring, where funding is landing, what headlines mean for growth, and how BioAI companies are thinking about commercialising what they’re building.
Each edition pulls from ongoing conversations with people doing the work day-to-day, as well as his own take on what’s hit headlines that month.
Joe recruits in this space day-to-day, and so often speaks from that vantage point. He spends most of his time inside these teams, hiring for them, speaking with founders and senior talent across the market. The aim isn’t to overstate where things are going, but to give a clear picture of what’s actually happening and why it matters if you’re hiring or looking to commercialise in BioAI.
🗓️ Events & Competitions
The best competitions, hackathons, and community challenges in AI x life sciences, curated weekly. Know something worth featuring? Reply and let us know.
More upcoming events:
BioHackathon Europe 2026 | November 9-13, Barcelona
ELIXIR’s annual international bioinformatics hackathon, running since 2018. 160+ participants, five days of collaborative coding on open bioinformatics infrastructure and tools. The call for project proposals opens March 16 and closes April 15 - so if you want to lead a project, that’s your window.
Thanks for reading!
💬 Get involved
We’re always looking to grow our community. If you’d like to get involved, contribute ideas or share something you’re building, fill out this form or reach out to me directly.
Connect With Us
Have questions or suggestions? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website