
ParameterizANI: AI for parameterization
Deep Dive | Edition 1


Welcome back to the deep dive, where we break down the AI tools and data reshaping how new drugs are discovered. In each edition, we speak directly with the teams behind these tools to explain what they solve, how they work and where they are going next.
In the weeks ahead, every Wednesday, we’ll be exploring novel tools like RFdiffusion 3, Germinal and ParametrizANI, and spotlighting standout companies like Elnora AI that are redefining how science gets done.
We spoke with Pablo Ricardo Arantes, the scientist behind ParametrizANI, an open-source project that reimagines one of the most painful bottlenecks in molecular simulations: parametrization.
After earning his PhD in Brazil and completing postdoctoral research in California under Professor Julia Palmo, Pablo joined EMS, Brazil’s largest pharmaceutical company, where he now helps lead the country’s first drug discovery team. Alongside that work, he set out to make high-accuracy parametrization fast, free, and accessible to anyone with a browser, combining neural network potentials with Google Colab notebooks to bring simulation power to anyone, anywhere.
Also, an important side note, the the paper was recently accepted for publication in JCIM (Journal of Chemical Information and Modeling). Well deserved Pablo and team!
🔴 The Problem
Accurately simulating a drug molecule’s behavior hinges on parametrization: defining how atoms and bonds behave inside a force field. Among the trickiest are dihedral angles, the rotatable bonds that shape how molecules fold and interact.
Traditionally, parametrizing dihedrals requires heavy-duty quantum mechanical calculations. They’re precise but slow, sometimes taking hours per molecule, and require supercomputers most labs simply don’t have. The process is also fairly cumbersome: older workflows often relied on genetic algorithms for fitting, which were complex and time-consuming to implement.
For Pablo, who encountered these roadblocks during his PhD and postdoc, the experience was both frustrating and inspiring. “Parameterization takes so much time. It’s boring, repetitive, and most people don’t have access to the supercomputers you need,” he told us. It was clear that if parametrization was going to keep pace with the rapid progress in drug discovery, it needed to be reimagined.
💡 The Idea
ParametrizANI was born from a simple thought: what if parametrization could run in the cloud?
Instead of relying on supercomputers, Pablo built a set of ready-to-use Jupyter notebooks on Google Colab. With them, any researcher or student can perform high-quality parametrization in a matter of minutes, no installation required.
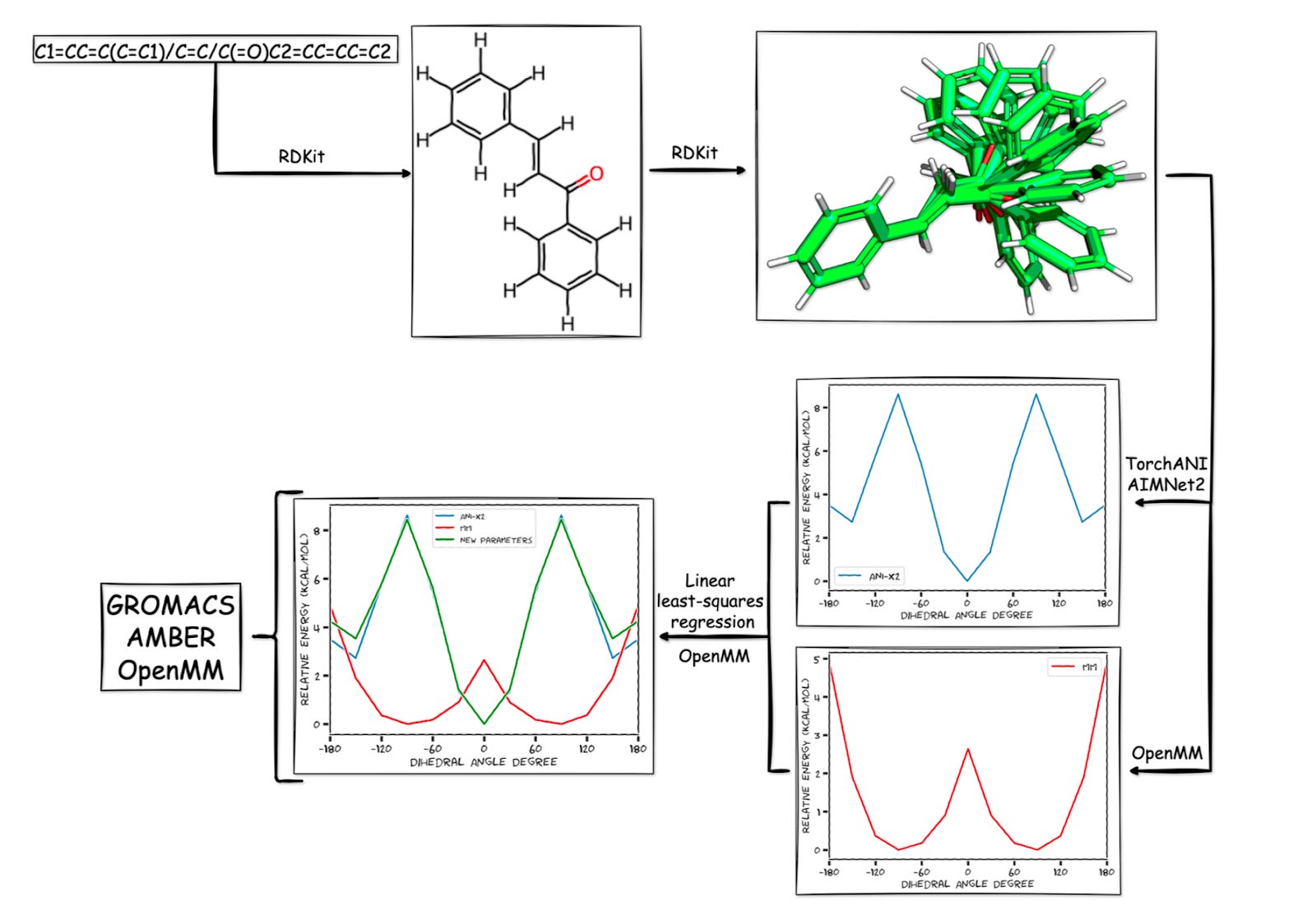
At the heart of the tool is TorchANI, a PyTorch-based deep learning framework that replicates quantum-level accuracy at a fraction of the cost. Trained on vast datasets of DFT calculations, TorchANI replaces the need to run new quantum mechanical simulations from scratch. Since its first release, Pablo has expanded ParametrizANI to support newer models like AIMNet2, MACE-OFF, and GFN2-xTB, ensuring the workflow stays current with the field’s rapid evolution.
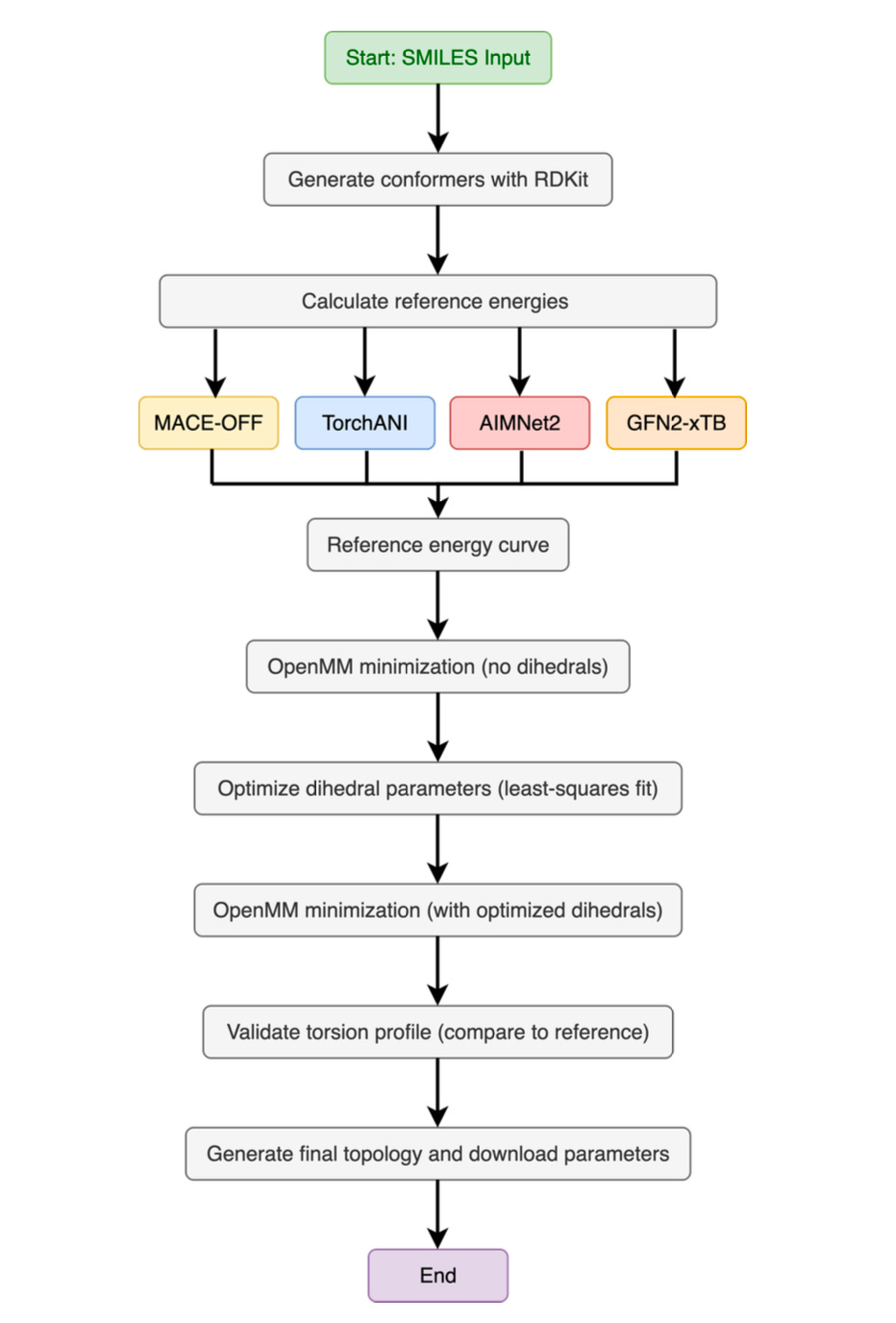
The result is a pipeline that can generate conformers, calculate reference energy profiles, fit dihedral parameters, and validate the results all inside a Colab notebook. What once demanded specialized hardware can now be done with a free Google account and a few clicks.
🔬 Why It’s Different
Plenty of parametrization tools exist but ParametrizANI stands out for how it lowers barriers to entry while keeping scientific rigor.
Accessibility: Runs entirely in Google Colab, requiring nothing more than a browser. No installs, no licenses, no HPC access.
Speed: Linear least-squares fitting replaces slower genetic algorithms, bringing runtimes down to just five minutes for a dihedral.
Flexibility: Compatible with GAFF and OpenFF force fields, with options to bring in external QM references or use integrated Psi4.
Education-ready: Transparent workflows and visualizations make it perfect for classrooms, helping students see each step without crashing local machines.
Evolving with the field: Pablo continuously updates the tool to incorporate new ML potentials like TorchANI 2, keeping pace with advances in the community.
In practice, that means a task that once excluded most researchers is now open to anyone, from large pharma R&D groups to students running their first simulations.
🔮 The Future
ParametrizANI is only at the start of its journey. Pablo is already working on packaging the workflow into a proper Python library installable via pip, making it even easier to plug into industrial drug discovery pipelines. He’s also pushing toward multi-dimensional dihedral parametrization, which could unlock new levels of accuracy for complex molecules. Exciting possibilities.
The long-term vision goes beyond small molecules. Future versions aim to handle larger biomolecular systems, including nucleic acids and metalloproteins, with potential integration into specialized tools like MCPB.py. Pablo also sees opportunities to incorporate advanced charge models, tackling another major bottleneck in force field preparation.
For him, the philosophy is pretty clear: “AI is never going to replace physical simulations but it can make them much faster. ParametrizANI is an example of that, we use AI to generate the parameters but the real insight still comes from the simulations.”
Now a part of Brazil’s first drug discovery team at EMS, Pablo is already testing this philosophy in practice. What makes it even more striking is that ParametrizANI has grown as a side project alongside his day job, reflecting both his industry insight and his commitment to making advanced tools freely accessible. The combination of speed, accessibility and continuous learning makes ParametrizANI a powerful companion in an industry where iteration speed is everything.
📄 Read the paper!
⚙️ Try the code out.
👨🔬 Get in touch with Pablo
Thanks for reading!
Did you find this newsletter insightful? Share it with a colleague!
Subscribe Now to stay at the forefront of AI in Life Science and keep up with this upcoming season of deep dives.
Connect With Us
Have questions or suggestions for our next deep dive? We’d love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website