
Pavia’s MOLECULE, Boston’s CryoPhold, and CNIO’s BinderFlow
Kiin Bio's Weekly Insights

Welcome back to your weekly dose of AI news for Life Science!
We’ve had some feedback from readers asking for more detail on the data behind the models we feature.
Starting this week, we’re adding a “What about the training data?” section to each article: covering what the model was trained on, where it came from, and how it was used.
What’s your biggest time sink along the drug discovery process?
MOLECULE: Learning how ligands shape protein behaviour, not just where they bind
A new study from the University of Pavia and Eni introduces MOLECULE, a deep learning framework that classifies ligands as orthosteric or allosteric by combining chemical structure with insights from protein motion.
Allosteric ligands are increasingly important in drug discovery, offering subtle and selective control over protein function. But predicting them is notoriously difficult. Most machine learning models rely on static features, while allostery is dynamic. Full molecular dynamics (MD) simulations help, but don’t scale.
MOLECULE bridges that gap. It learns from short MD simulations of how key binding pocket residues move in response to a ligand, then combines that with a Morgan fingerprint of the ligand. The result is a model that infers not just where a ligand binds, but how it changes protein behaviour. Interesting take.
To make this usable in screening, the team also trained an imputer that approximates the MD signal using only the ligand fingerprint. This enables predictions when simulation data is unavailable or impractical.

Applications and Insights
1. Combines structural and dynamic information
MOLECULE processes both a ligand fingerprint and the time-series motion of six key binding site residues. This dual-input setup improves test accuracy to 92 percent, up from 85 percent using structure alone. Nice jump.
2. Predicts without full simulations
The imputer reconstructs a latent dynamics signal from the ligand fingerprint alone. Even without MD data, the classifier maintains 89 percent accuracy, enabling fast and scalable use.
3. Prioritises high-confidence predictions
The model includes a confidence estimation mechanism, allowing researchers to filter results based on reliability. This is especially useful for avoiding false positives in high-throughput settings.
4. General and extensible framework
MOLECULE is trained on kinase-ligand complexes but is generalisable to other protein families. The full codebase is open source and designed to be adapted for broader classification tasks in drug discovery.
What about training data?
MOLECULE doesn’t rely on large pretrained models. Instead, it learns functional ligand effects from paired structure and simulation data:
Each input consists of a kinase–ligand complex from the KLIFS database, paired with a short molecular dynamics simulation that captures how six pocket residues move in response to binding.
These dynamics are encoded as time-series features and combined with a standard Morgan fingerprint of the ligand. The model is trained to classify each ligand as orthosteric or allosteric based on this combined signal.
To make the tool usable without running simulations, a second model learns to impute the dynamics signal from the ligand fingerprint alone — enabling fast screening without MD.
All data sources are public, and the full framework is available open-source for retraining or extension to other protein families.
I thought this was cool because it reflects a shift toward functionally-aware learning in molecular modelling. Instead of predicting what binds where, MOLECULE predicts how a ligand affects a protein and does so with or without simulations.
It is rare to see this kind of balance between mechanistic insight and practical usability. MOLECULE hits both, which makes it a valuable tool for anyone working on allostery, kinase inhibitors, or smart screening strategies.
📄Check out the paper!
⚙️Try it out the code.
CryoPhold: Modelling protein dynamics with AlphaFold, cryo-EM, and machine learning
A new framework from Washington University and Boston University introduces CryoPhold, a modular pipeline that combines structure prediction, cryo-EM, simulation, and machine learning into a single workflow for uncovering protein dynamics.
Most tools return a static structure, but proteins aren’t static. CryoPhold generates ensembles, maps energy landscapes, and identifies slow, functional motions using only sequence and cryo-EM data. This opens new ways to study how proteins work and how their states shift with mutation or ligand binding.
The team tested CryoPhold on two systems: the glycine transporter GlyT1, a target in schizophrenia, and oncogenic BRAF mutants, which drive melanoma. In both cases, it captured population shifts and revealed regulatory hotspots.

Applications and Insights
1. Extracts protein dynamics from cryo-EM
CryoPhold uses cryo-EM maps not just for fitting, but to guide Bayesian ensemble refinement. It recovers metastable states and variability, even from low-resolution or averaged data.
2. Resolves functional states in GlyT1
With just the apo structure and cryo-EM, CryoPhold captured the inward > occluded > outward equilibrium and identified Y62, whose flipping drives inhibitor binding. It also recovered flexible loops missing from crystal structures.
3. Explains BRAF mutation effects
Simulations seeded from CryoPhold ensembles showed V600E and V600K mutations shift BRAF toward active conformations, altering the DFG motif, αC-helix, and activation loop. These changes weren’t visible in the cryo-EM map but emerged from the ensemble.
4. Learns slow, meaningful features
CryoPhold extracts features like dihedrals and distances directly from MD. These can support Markov models, enhanced sampling, or allosteric drug design.
What about the training data?
CryoPhold doesn’t use a traditional pretraining dataset. Instead, it builds its own system-specific dataset by combining predictions, maps and simulations:
Structure inputs come from 50 AlphaFold2 models per target, aligned to experimental cryo-EM maps. These structure/map pairs, along with 100 ns molecular dynamics simulations, are fed into CryoPhold to capture the protein’s conformational transitions.
Learning happens post hoc using unsupervised methods, specifically time-lagged autoencoders and slow feature analysis, trained directly on the MD trajectories.
All inputs are public, and the entire pipeline is fully reproducible using open-source tools.
I thought this was cool because it connects four separate domains: AlphaFold, cryo-EM, simulation, and ML into one pipeline. It doesn’t rely on massive pretrained models, just what you can generate from sequence and structure.
CryoPhold is especially useful when you want to go beyond static predictions and explore how proteins move, how their motions shift, and what features actually govern those transitions. A strong tool for mechanism discovery, screening, and engineering.
📄Check out the paper!
⚙️Try it out the code.
BinderFlow: Parallelised protein binder design for any lab with a GPU
A new framework from the Spanish National Cancer Research Centre introduces BinderFlow, a modular pipeline for designing protein binders with deep learning. Built on top of RosettaFold Diffusion (RFD), it automates and speeds up the design process, making it far more accessible to labs without specialised infrastructure or deep protein design expertise.
BinderFlow runs the full binder design process: backbone generation, sequence assignment (via pMPNN), and scoring with AlphaFold2 and PyRosetta in small, independent batches. Each batch can run on a single GPU, enabling high-throughput campaigns without queue bottlenecks. A real-time dashboard called BFmonitor tracks progress and lets users stop runs once enough hits are found.
By splitting tasks across GPUs and letting users monitor results live, BinderFlow offers a faster, more flexible way to generate binders and helps researchers avoid compute waste or redundant designs.
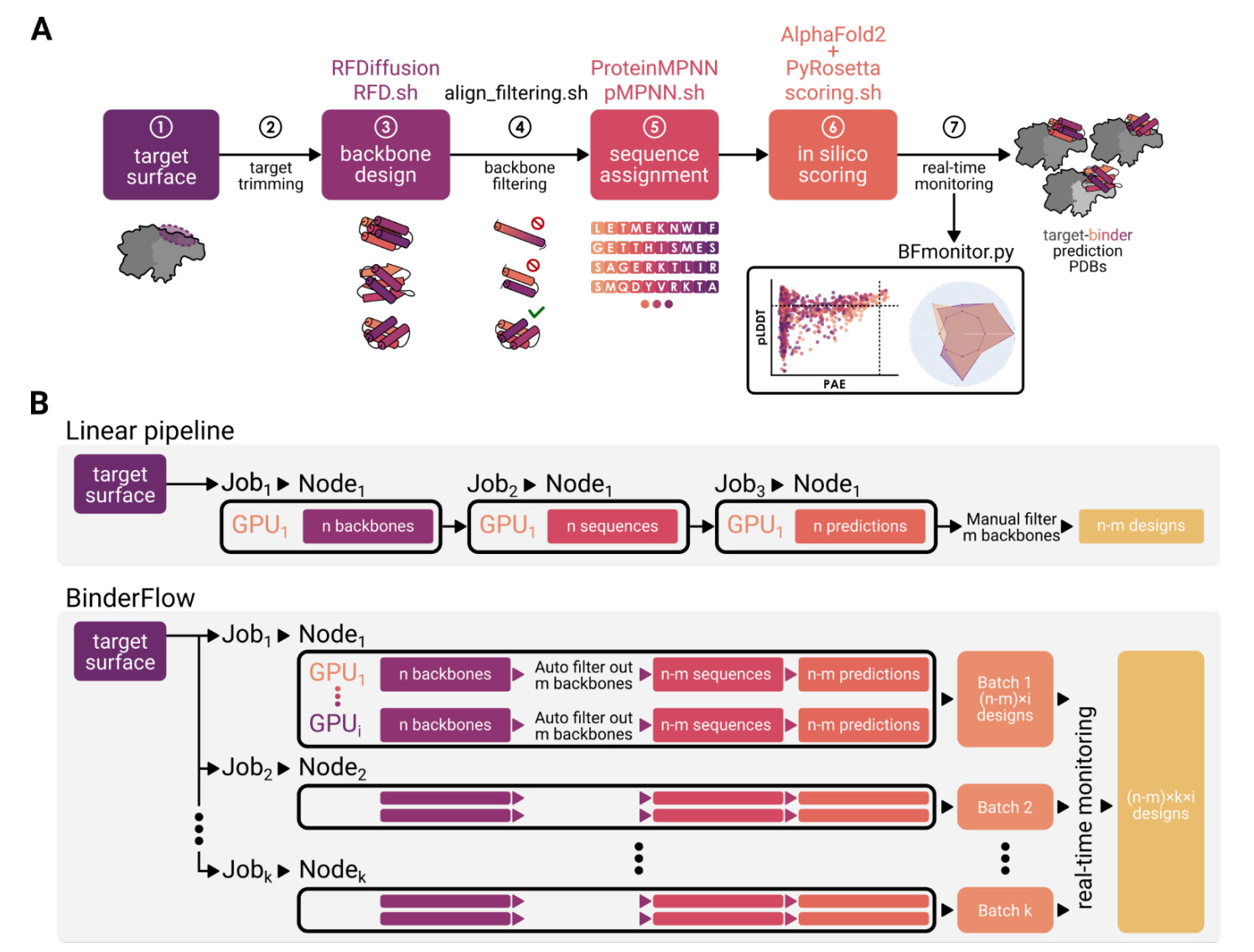
Applications and Insights
1. Fast, flexible, GPU-ready
BinderFlow breaks large design jobs into small, parallel tasks that run independently on available GPUs. This allows users to repurpose idle hardware. In one benchmark, it delivered 24 high-quality binders in under 15 hours — with real-time updates throughout.
2. Smart refinement options
Two optional modules, Partial Diffusion and Sequence Diversity, help improve quality without more compute. The former perturbs existing backbones to explore local structure space; the latter samples multiple sequences per backbone for better complementarity.
3. Less waste, fewer bottlenecks
With live scoring and filtering, users can stop early once enough strong hits are found avoiding wasted runs and reducing downstream experimental effort.
4. Designed for actual researchers
The visual dashboard supports filtering, ranking, and export of sequences and structures. It even includes a built-in codon optimiser for DNA ordering. BinderFlow runs on any SLURM-based system, from personal GPUs to full clusters.
What about the training data?
BinderFlow doesn’t train new models, it orchestrates existing pretrained tools like RFD, pMPNN, AlphaFold2, and PyRosetta in a reproducible workflow.
All design and benchmarking used publicly available structures from the AlphaFold Protein Structure Database (AFDB).
The entire pipeline runs on standard SLURM-based systems using consumer-grade GPUs, so no specialised compute is needed.
I thought this was cool because it solves a real usability problem. We’ve seen huge progress in binder design models but many labs still find them hard to run. BinderFlow doesn’t invent new models, it just makes the existing ones usable.
That kind of engineering deserves more attention, especially in a field as fast-moving as protein design. For labs who want to get into this space without a full redesign of their compute setup, this is a seriously practical step forward.
📄Check out the paper!
⚙️Try it out the code.
Thanks for reading!
Did you find this newsletter insightful? Share it with a colleague!
Subscribe Now to stay at the forefront of AI in Life Science.
Connect With Us
Have questions or suggestions? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website