
🧩 Reconstructing the Fold: How Bioinspired Variables Fix a 20-Year Problem
Deep Dive | Edition 8


Welcome back to the deep dive, where we break down the AI tools and data reshaping how new drugs are discovered. In each edition, we speak directly with the teams behind these tools to explain what they solve, how they work and where they are going next.
This time we spoke with Simone Aureli, a postdoctoral researcher in Francesco Gervasio’s group at the University of Geneva, about a new strategy for modelling protein folding. While AlphaFold solved the problem of predicting protein structures, it left open the question of how proteins actually fold: the thermodynamics, the intermediates, and the pathways. That is where the Gervasio lab’s hard work comes in.
🔴 The Problem
Protein folding remains one of the hardest challenges in molecular biophysics. Folding happens on the microsecond to millisecond timescale, far beyond what most molecular dynamics (MD) simulations can reach. Specialised hardware like the Anton supercomputer has generated landmark folding trajectories but access is limited and the cost makes it fairly prohibitive.
Enhanced sampling methods, such as metadynamics or OneOPES, provide an alternative. But their performance hinges on the choice of collective variables (CVs), the reduced coordinates that describe folding. Poor CVs lump together distinct states or miss intermediates, making simulations misleading. This degeneracy problem has meant that folding pathways often remain unresolved, especially in systems larger than a handful of residues.
💡 The Idea
The Geneva team set out to design bioinspired CVs that explicitly encode the microscopic details of folding.
They developed two complementary CVs:
sHB, which tracks hydrogen bonds with angular detail and distinguishes protein-protein from protein-water interactions.
sSC, which captures side-chain packing, explicitly including both native and non-native contacts.
Together, these CVs span both microscopic and mesoscale features of folding. Unlike generic distance-based CVs, they are constructed bottom-up from short unbiased simulations of folded and unfolded states, then filtered with a linear discriminant analysis style criterion.
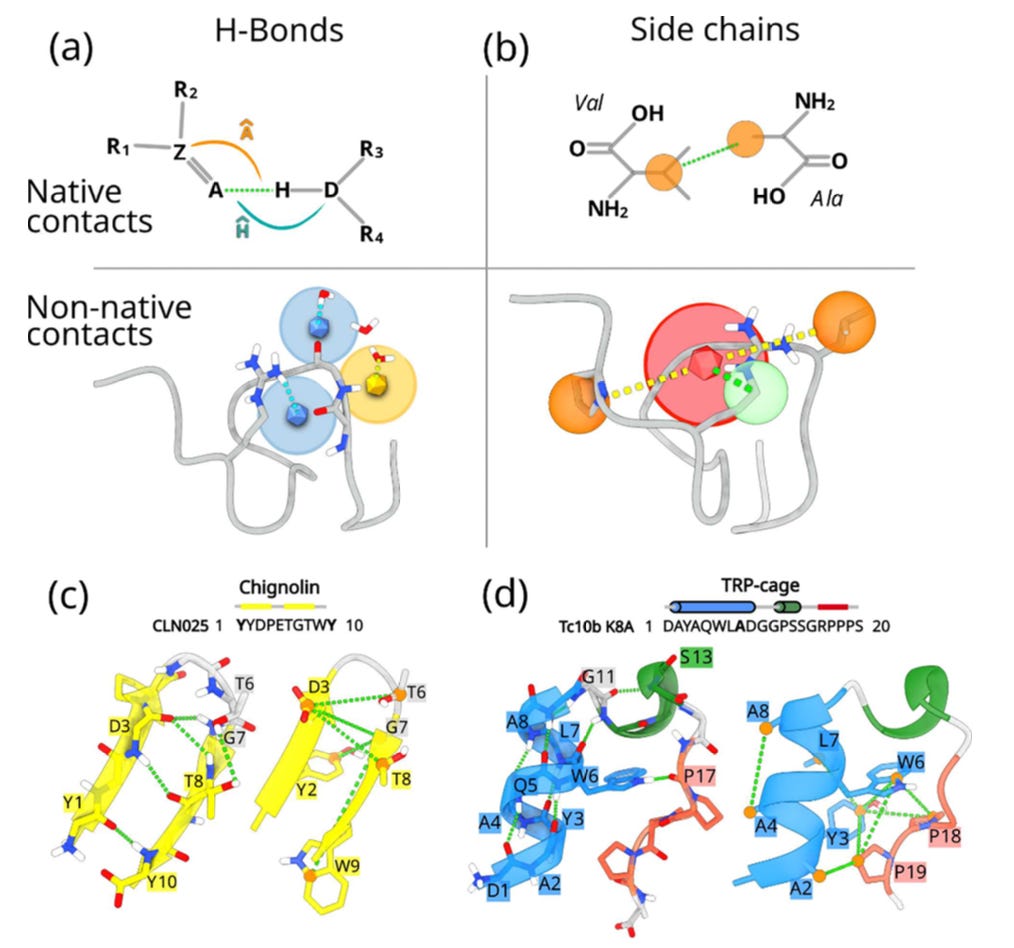
A central aspect of their design is that the CVs do not only reward the formation of correct contacts and hydrogen bonds; they also actively penalise competing alternatives. In other words, nearby residues are discouraged from forming “almost right” interactions, which could otherwise create hysteresis and obscure the folding pathway. By penalising these nearly correct but misleading states, the CVs sharpen the description of the folding landscape, producing a cleaner separation of relevant states. This idea is a crucial difference compared to more permissive approaches and significantly improves the interpretability of the simulations.
📊 What about the training data?
The training signal did not come from large curated databases but from reference MD trajectories generated by the group itself.
For Chignolin: three 100 μs unbiased simulations with 76 folding transitions.
For Trp-cage: one 200 μs unbiased simulation with 26 transitions.
These trajectories provided the ground truth for validating enhanced sampling runs with the new CVs.
All the data, input files, and scripts have been openly released on GitHub and PLUMED-NEST so that anyone can reproduce the results or build upon them.
🔬 Why It’s Different
Unlike most folding CVs, which rely on global measures like RMSD or radius of gyration, the Geneva team’s approach is built on explicit microscopic features.
Explicit solvent and hydrogen bonds: By encoding protein–protein versus protein-water hydrogen bonding, the method captures water’s role in folding.
Native and non-native contacts: By subtracting non-native from native interactions, the CVs distinguish unfolded, misfolded, and folded states without degeneracy.
Complementary resolution: sHB captures fine-grained hydrogen bonding, while sSC encodes the broader hydrophobic collapse, together spanning the folding funnel.
Validated landscapes: The method reproduced folding free energies for Chignolin and Trp-cage with striking accuracy, resolving elusive intermediates such as the wet and dry molten globule.
Transparent and portable: The approach is implemented as an open Python script that outputs ready-to-use PLUMED files.
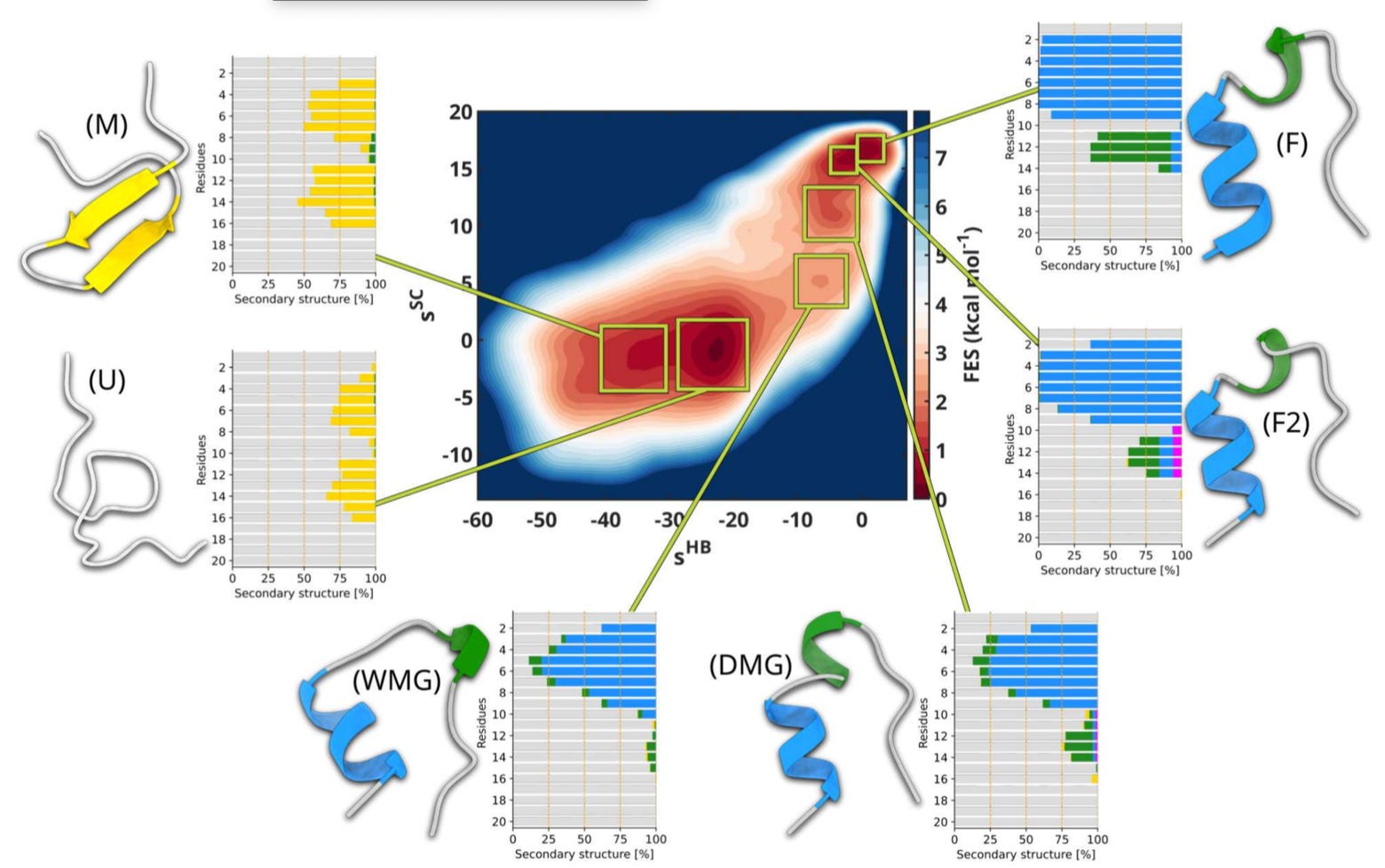
🔮 The Future
The current CV strategy works well for mini-proteins like Chignolin and Trp-cage, but the team is already moving into more complex systems. They are applying it to GPCR signalling proteins such as beta-arrestin 1 and to RNA filaments, where ions are essential for stability, and they see membrane proteins as the next major frontier, since these will require accounting for the variance introduced by phospholipids and cholesterol.
Alongside these applications, they are committed to making all trajectories, input files, and training sets openly available, so that folding simulations can become not just predictive but reproducible and trustworthy tools for exploring the full landscape of biomolecular change.
📄 Read the paper!
⚙️ Try it out on GitHub or PLUMED-NEST.
👨🔬 Get in touch with Simone.
Thanks for reading!
Did you find this newsletter insightful? Share it with a colleague!
Subscribe now to stay at the forefront of AI in Life Science and keep up with this upcoming season of deep dives.
Connect With Us
Have questions or suggestions for our next deep dive? We’d love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website