
RosettaCommons' RF3, MIT's Antibiotic Designer and Helmholtz's TPM Model
Kiin Bio's Weekly Insights

Welcome back to your weekly dose of AI news for Life Science!
Reminder: Our Tools Atlas database contains the codes for the open-source tools mentioned in this issue and all previous newsletters!
Is there any task that you find drains your time in the drug discovery process?
Let us know what is and we’ll send you open source tools specific to that issue.
RosettaFold3: Unified structure prediction for proteins, ligands, RNA
Right now, predicting how proteins bind to ligands or RNAs means juggling around a load of separate tools. Not ideal. Some are accurate, some are open, but you can rarely find a tool that is both. A new open-source release from the Rosetta team has just changed that.
The Rosetta Commons group has introduced RosettaFold3 (RF3), a structure prediction model that handles proteins, peptides, RNA, and ligand complexes. It is trained end-to-end on over one million examples, supports atomic-level templates and constraints, running on an open and modular framework called AtomWorks.

Applications and Insights:
Improved ligand chirality prediction
RF3 increases ligand chirality accuracy to 88% , outperforming AlphaFold3 at 84% and Boltz-2 at 76%. Chirality plays a key role in drug activity, so these improvements help ensure more realistic modelling of small molecule interactions.
More accurate protein-ligand interface modelling
Adding atomic-level constraints improves protein-ligand interface accuracy from 0.821 to 0.890. This makes RF3 well suited for docking workflows and drug discovery tasks that rely on precise interface geometry.
Robust handling of noncanonical peptides
RF3 recovers 86 percent of chiral centres in nonstandard peptides and reaches a 1.74 Å RMSD, supporting accurate modelling of modified or synthetic peptide structures often used in therapeutic design.
Works across multiple input types
The model handles proteins, ligands, RNA, antibodies, and docking targets within the same system. This simplifies prediction pipelines and saves time by avoiding the need to switch tools for each molecular format.
AtomWorks makes this possible by standardising structural biology inputs into much cleaner, extensible formats. So it processes proteins, ligands, and nucleic acids as modular AtomArray objects and then applies transformations similar to how image models use torchvision. For researchers building biomolecular models, this removes the need to reinvent every single pipeline from scratch, which has previously been the case!
I thought this was cool because it’s a proper shift toward better infrastructure for molecular modelling. AtomWorks turns atom-level data into something you can actually reason about and build on. That kind of modularity is what lets science scale, so it will be interesting to see how this plays out long term.
📄Check out the paper!
⚙️Try the code.
Antibiotic Designer: Multi-objective molecule generation for safe, potent antibiotics
Antibiotic resistance is rising. New antibiotics are struggling to catch up.
Part of the problem here is that designing safe, effective small molecules is still quite a slow and compartmentalised process. This new generative pipeline tries to fix that by marrying chemistry and biology in the design phase, not just in experimental screening.
Researchers at MIT have released a deep learning system that generates antibiotic candidates from nothing and filters them based on potency, toxicity, and predicted mechanism of action. In my opinion, this looks like one of the most in depth and comprehensive AI integrated antibiotic discovery approaches to date.

Applications and Insights:
1. Designed molecules with real-world potency
The model generated two promising candidates, DN1 and NG1. DN1 showed strong activity against Staphylococcus aureus, and NG1 was effective against drug-resistant Neisseria gonorrhoeae, including strains where other treatments fail.
2. Toxicity-aware generation with chemprop
A built-in toxicity predictor helped screen out compounds that might harm human cells, improving the therapeutic index and saving time and resources on low-probability candidates.
3. Biology-guided design across various data types
Unlike SMILES-only models, this system incorporated bacterial morphology, transcriptomics, and cryo-EM data to help steer generation toward preferred mechanisms like membrane disruption or cell wall stress.
4. Validated predictions across scales
Designed compounds were validated with transcriptomics, SHAP analysis, and imaging. They showed clean phenotypic profiles and targeted known antibacterial pathways while avoiding human off-target effects.
What stands out here is how much real biology the model considers. It treats molecules not just as lone standing strings to optimise but as agents with proper phenotypic consequences. That shift from chemistry-only to biology-aware design could be key to translating AI predictions into drugs that actually work.
I thought this was cool because it shows how far generative design can actually go when you bring in real experimental context. Instead of chasing accuracy on toy benchmarks, this work brings together generation, prediction, and wet-lab feedback into a single loop. Which I think is exactly how discovery should look.
📄Check out the paper!
⚙️Try out the code.
Target Preference Maps (TPM): 3D atom preference maps for structure-guided drug design
Designing small molecules still feels like guesswork.
Even with high-resolution structures and large virtual screens, most computational tools don’t directly tell you what kinds of atoms a binding pocket wants. They focus on whether a compound fits, not how to make it better.
This new model from Helmholtz Munich flips that on its head. It learns the “preferences” of binding pockets at the atomic level and generates 3D maps of what kinds of atoms are most likely to bind where. It’s called the Target Preference Map (TPM), and it is an incredibly intuitive tool for turning structural biology into actionable chemistry.
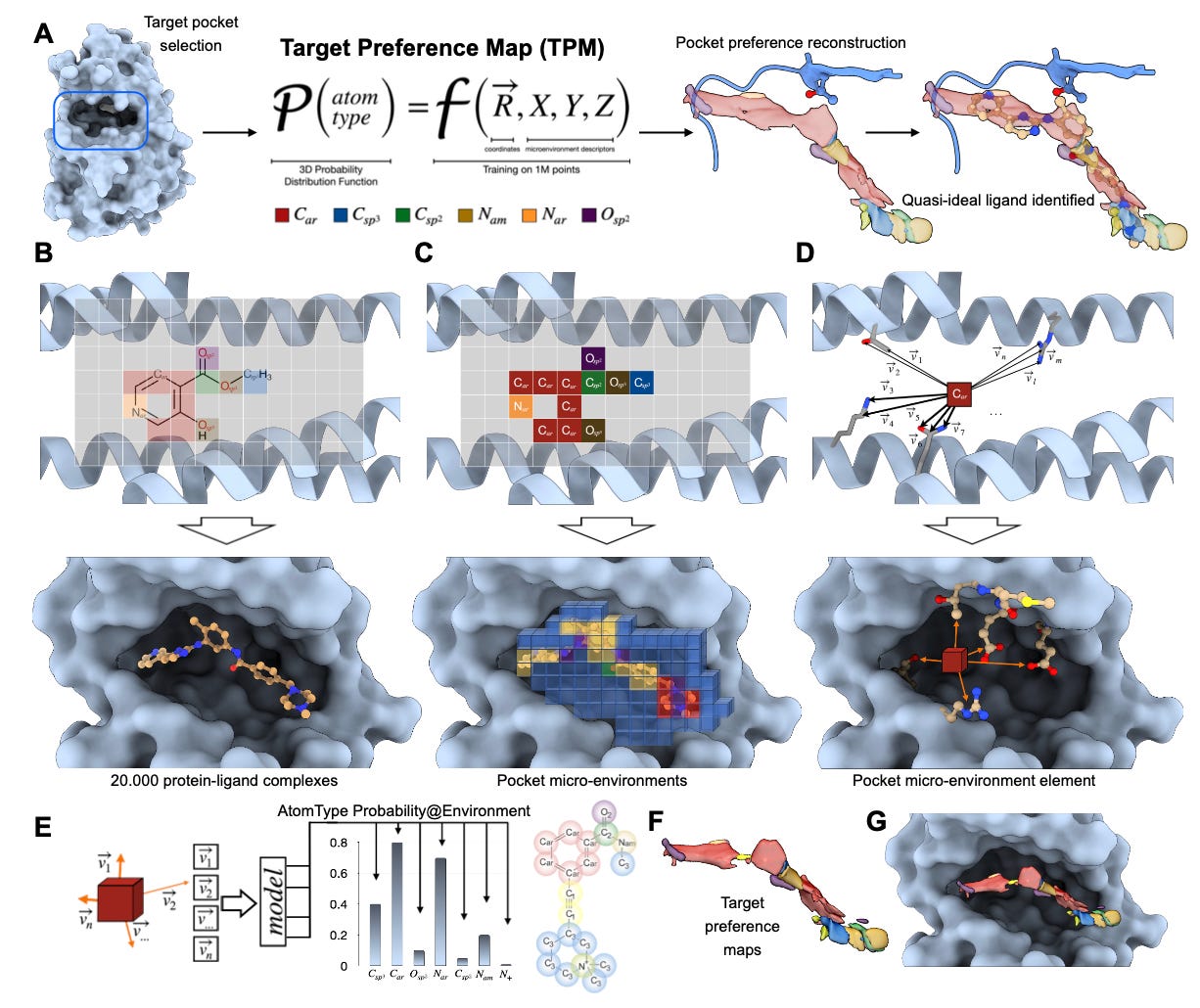
Applications and Insights:
1. Atomic resolution guidance
Predicts local preferences for 15 atom types with over 85% classification accuracy, based only on binding pocket structure.
2. Improves lead optimisation
In a real-world test on PEX14 inhibitors, TPM-guided edits delivered a 10× gain in potency (from 30 μM to 3 μM)
3. Generalises to unseen targets
Achieved 83% AUC on entirely novel protein families and correctly flagged Zn-binding pockets it was never even trained on.
4. Visual and interpretable
Outputs are 3D density maps readable in PyMOL or Chimera, letting chemists see precisely where to add or modify atoms.
I thought this was cool because TPM changes how we approach and think about pocket-based design.
Instead of testing molecules and hoping one fits, you start from the biology and ask: what kind of chemistry would this pocket prefer? So really there’s a shift from evaluation to generation and one that makes medicinal chemistry more grounded in structural logic, not just ligand similarity.
TPM’s interpretability is what I found especially valuable. The fact that you can open the outputs in PyMOL and literally see the suggested regions for fluorines, sulfurs, or aromatic nitrogens gives medicinal chemists a real and tangible starting point. In addition, since it doesn't rely on knowing the ligand ahead of time, it’s not just useful for optimisation. Really, it’s useful for designing from scratch, which is huge.
These kinds of intuitive, structure-native tools do feel like the next layer of infrastructure we need to speed up rational drug discovery.
Check out the paper!
The code is planned for release following approval of the manuscript.
Thanks for reading!
Did you find this newsletter insightful? Share it with a colleague!
Subscribe Now to stay at the forefront of AI in Life Science.
Connect With Us
Have questions or suggestions? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website