
🧪 Rowan: Computational Chemistry Without the Code
Deep Dive | Edition 17

Welcome back to the deep dive, where we break down the AI tools and data reshaping how new drugs are discovered. In each edition, we speak directly with the teams behind these tools to explain what they solve, how they work and where they are going next.
Keeping up with AI x life science news can get exhausting.
It’s scattered across LinkedIn, X, Substack, arXiv, Slack, newsletters... and you still somehow miss the things that actually matter. Too much noise, not enough signal.
We’re building something to fix that: a smarter, more powerful way to stay on top of what’s actually relevant to you.
But we want to build it with you, not just for you. Take 2 minutes to tell us what’s missing. What you share will directly shape what we build, and you’ll be the first to benefit from it.
Today we’re looking at Rowan, a Boston-based startup of six that’s building a web-based computational chemistry platform that lets medicinal chemists run advanced modeling workflows directly, without needing to manage scripts, infrastructure, or specialist software.. We spoke with co-founder Corin Wagen, an experimental organic chemist turned computational entrepreneur, about why the gap between medicinal chemists and computational tools has persisted for so long, and what it takes to close it.
“There are all these really smart, really talented chemists and scientists who are just not able to use computation to help them out. You always have to ask somebody else to do it. There’s all these artificial barriers.”
— Corin Wagen, Co-founder, Rowan

🔴 The Problem
Computational chemistry has a usability problem.
The tools exist. You can predict binding affinities, generate conformers, run molecular dynamics, dock compounds into protein structures. But actually using any of this typically requires programming expertise, command-line fluency, access to the right hardware, and the patience to stitch together a dozen different software packages that were never designed to work together.
For most medicinal chemists, the people actually deciding which compounds to synthesise next, this means going through a computational chemist every time they want to run something. That handoff slows everything down. It creates bottlenecks, introduces miscommunication, and means that computation gets used selectively rather than routinely.
The result: most drug discovery teams are making synthesis decisions with less computational insight than they could be: not because the science isn’t there, but because the software gets in the way.
“You should be a chemist to use Rowan, but you shouldn’t need to be a programmer.”
💡 The Idea
Rowan’s answer is a web-based platform that organises computational chemistry into workflows, data in, data out, aligned to how scientists actually think about experiments.
You have a compound and want to know how soluble it will be? That’s a workflow. You have a binding pose and want to screen 100 analogues? That’s a workflow. Under the hood it might be an ML model, a physics simulation, or a database lookup, but the scientist doesn’t need to care about the plumbing.
The founding team brings a deliberate mix: machine learning, software engineering (ex-Meta), quantum chemistry, product and business, and experimental organic chemistry. Wagen sees that breadth as essential. “A lot of times the best ideas in chemistry come from people who’ve journeyed outside chemistry and then bring back new ideas.”
The platform spans workflows from hit discovery to candidate selection, ligand-based methods (ML potentials, rapid quantum chemistry, conformers, reactivity, spectra) and, increasingly, structure-based drug discovery (docking, co-folding, molecular dynamics, and now free energy perturbation). Scientists pick what they need. Rowan doesn’t prescribe a single workflow.
Critically, Rowan is designed to fit into existing scientific software stacks. Teams can use it through the browser, through Python, or as part of agentic and automated pipelines, without replacing the tools they already rely on.
“There’s a lot more smart people outside Rowan than inside Rowan. We don’t need to own the whole thing.”
⚙️ The FEP Release
The headline addition is free energy perturbation (FEP), the gold-standard physics-based method for predicting how binding affinity changes across a series of related compounds. If you’re optimising a lead and need to decide which of 100 analogues to actually synthesise, FEP tells you which ones are likely to bind better and which are duds, before you spend time and money making them.
FEP has been around for decades, but two things have kept it out of mainstream medicinal chemistry workflows: it’s expensive (historically around 10 GPU hours per compound) and it’s complicated to set up and run.
Rowan partnered with Forrest York and the open-source TMD engine (originally from Relay Therapeutics) to tackle both. The engineering improvements are dramatic: default settings run approximately 10 minutes per leg, with the potential to reach 1-2 minutes per leg with adjusted settings. That’s a roughly 60x speedup over the literature standard.
The speed comes from low-level optimisation and an algorithmic trick called local resampling, where instead of simulating the entire protein, the calculation focuses on the immediate neighbourhood around the ligand. “If you naively try to do that, it works very poorly,” Wagen explains. “But it turns out if you very cleverly try to do that, it works very well.”
The end-to-end workflow can be run without any coding. Three steps: prepare your poses using analogue docking, build a perturbation graph showing which ligands to compare, and run the FEP calculations on cloud GPUs. Results stream back to your browser in real time.

📊 The Trade-Off
The honest question: how accurate is it?
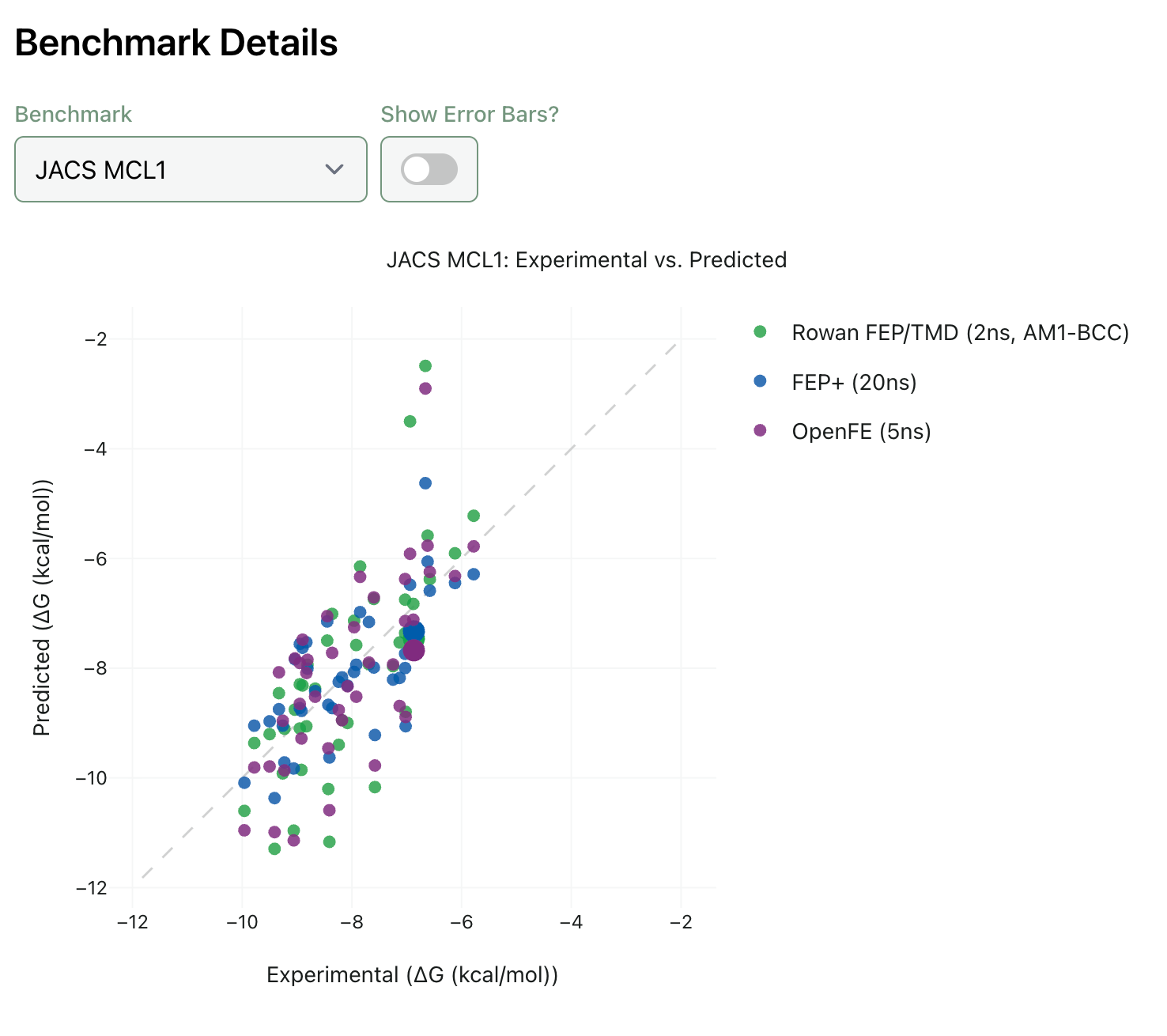
Rowan’s benchmarks show a mean absolute error of approximately 1.3 kcal/mol, compared to Schrödinger’s ~0.8 kcal/mol. The gap comes from two places: the speed-optimising approximations and the use of open force fields rather than Schrödinger’s proprietary ones.
Wagen is transparent about this. “Our benchmarks are a little bit worse than Schrödinger’s. They have amazing force fields. They’re so good at force fields.”
But the metric that matters most in practice isn’t absolute energy prediction, it’s ranking. If FEP correctly tells you which compounds will bind better and which won’t, you’ve saved synthesis cycles regardless of whether the exact energy numbers are perfect. And on ranking, the performance gap narrows considerably.
“If compounds are predicted to bind terribly, unless you really don’t understand the binding mode, they’re almost always bad binders. We have a lot of benchmark data showing this.”
The calculus is straightforward: if you can run FEP on 10–100x more compounds than you can synthesise, at a fraction of the cost and time, slightly wider error bars are a trade-off most teams will take.
Rowan publishes its benchmark data openly: you can explore the full results at benchmarks.rowansci.com.
🔬 Why It’s Different
Speed changes behaviour. At 10 GPU hours per compound, FEP is something you run occasionally on high-value decisions. At 10 minutes, it becomes routine, something you run on every idea before committing to synthesis. That shift from selective to systematic is the real unlock.
No code, no setup. Most FEP implementations require protein preparation scripts, force field configuration, graph construction code, and hardware management. Rowan handles all of this in three browser-based workflows, while also providing API access for teams that want to automate and run FEP at scale.
Open infrastructure, not a black box. Built on the open-source TMD engine and open force fields. Benchmarks are published. The platform is designed to complement existing tools via API, not replace entire workflows.
Built for the medicinal chemist. Rowan is built to put computational insight directly in the hands of medicinal chemists, while still remaining useful to computational and platform teams.
💊 Who It’s For
Rowan’s primary customers are small-to-medium biotechs and pharma departments that don’t have large internal computational tooling teams. The philosophy is to complement, not compete.
“We often work with companies where they’re small enough that they don’t have anybody who they can ask to do the work that we do for them,” Wagen says.
The platform recently passed 10,000 users and has generated over 40 publications. Pricing for FEP runs at approximately $5-10 per edge for platform customers, or $25 per ligand through a managed fee-for-service option where a Rowan scientist handles the analysis.
🔮 The Future
The near-term goal is integration into fast-moving drug discovery programmes through active pilots. The dream: automated nightly runs from SMILES strings to predicted binding affinities, with a digest landing in your inbox each morning showing which AI-generated analogues are worth pursuing.
“I suspect that the dream is that we can just blindly put SMILES in, go all the way to binding affinities, and that runs every single night.”
That dream is not yet reality. Fully automated structure-based modeling is still not trivial, particularly in pose preparation and graph construction, and Wagen is candid that the final stretch depends on solving messy, program-specific edge cases and learning from their early FEP customers. Beyond automation, the roadmap includes custom force-field fitting for client compound series (to close the accuracy gap),, pre-FEP triage tools, and continued speed optimisation targeting under one minute per compound for large libraries.
“I hope this doesn’t take us a few years. I hope this can happen in 2026 for Rowan.

👨🔬 Get in touch with Corin
📺 Watch the FEP walkthrough on YouTube.
📄 Read the FEP core concepts explainer.
Thanks for reading Kiin Bio Weekly!
💬 Get involved
We’re always looking to grow our community. If you’d like to get involved, contribute ideas or share something you’re building, fill out this form or reach out to me directly.
Subscribe now to stay at the forefront of AI in Life Science and keep up with this upcoming season of deep dives.
Connect With Us
Have questions on this or suggestions for our next deep dive? We’d love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website
An interesting read. And I've went through rowan and they are actually doing pretty good job. But I've a question why they are not collaborating or opting for academics people as I'll be mostly helpful for academics people I feel. And there's also tamarind bio how rowan going to keep up healthy competition with them?? Thanks for sharing!!!!!