
🧬 scConcept: Why Your Cell Embeddings Don't Work
Deep Dive | Edition 20

Welcome back to the deep dive, where we break down the AI tools and data reshaping how new drugs are discovered. In each edition, we speak directly with the teams behind these tools to explain what they solve, how they work and where they are going next.
We know how hard science is. That’s why we built the Pioneer Programme.
We’re selecting academic and nonprofit research teams to get one year of free access to our drug discovery platform plus hands-on support from our science team. If your research bottleneck isn’t data but connecting the findings you already have, this is for you.
No cost. No data transfer. All IP stays with your institution. Applications close August, cohort starts September.
Single-cell biology has spent the last few years borrowing ideas from language models. Several groups have adapted transformer architectures to gene expression data, including scGPT and Geneformer, both of which pretrain on massive single-cell datasets using masked modeling objectives.
Transformers. Pretraining. Big datasets. It all felt like progress. But if you ask most practitioners what they actually use day to day, the answer is still pretty modest: embeddings that work, transfer across datasets, and do not collapse when the technology changes.
That tension is what motivated scConcept, a new foundation model for single-cell transcriptomics that steps away from gene reconstruction and instead asks a simpler, more explicit question: do these genes come from the same cell?
We spoke with Mojtaba Bahrami, in Fabian Theis’ lab at Helmholtz Munich, part of the the team behind scConcept about why the field needed a reset, what contrastive learning brings to biology, and why the future may be less about bigger models and more about better representations
🔴 The Problem
If you zoom out, most single-cell foundation models share the same basic idea. Treat genes like words. Mask some of them. Ask the model to predict what’s missing. This strategy mirrors masked language modelling introduced in BERT, where models learn by predicting missing tokens in a sentence. It works well enough on paper, but something feels off once you start using the embeddings downstream.
As Mojtaba Bahrami put it when we spoke, “In language models, training and inference are basically the same task. You predict the next token. But in single-cell, no one actually cares about predicting masked genes. People care about the embedding.”
That mismatch matters. Masked gene prediction optimises the wrong thing. The model gets good at reconstructing counts, but the cell-level representation becomes an afterthought. Most approaches average learned gene embeddings and hope for the best. This may work depending on the downstream question but often it doesn’t. If the question can not be simply answered by looking at gene level information and needs understanding higher level biological processes going on in the cell, then we have a problem.
The cracks show up quickly. Simple methods like PCA or VAEs still outperform large models on tasks like cell type annotation. Benchmarking efforts such as the scIB framework have shown that classical integration methods can remain highly competitive across datasets. Spatial assays break embeddings entirely. New technologies shift the latent space so much that “foundation” starts to feel like marketing rather than reality.
💡 The Idea
Here comes scConcept. Instead of asking the model to reconstruct genes, it asks something more aligned with how the actual biology is: What is the identity of a cell given a partial set of its gene expression. In other words, can we identify the biological processes that make up the identity of a cell consistently through looking at different views of its transcriptome profile?
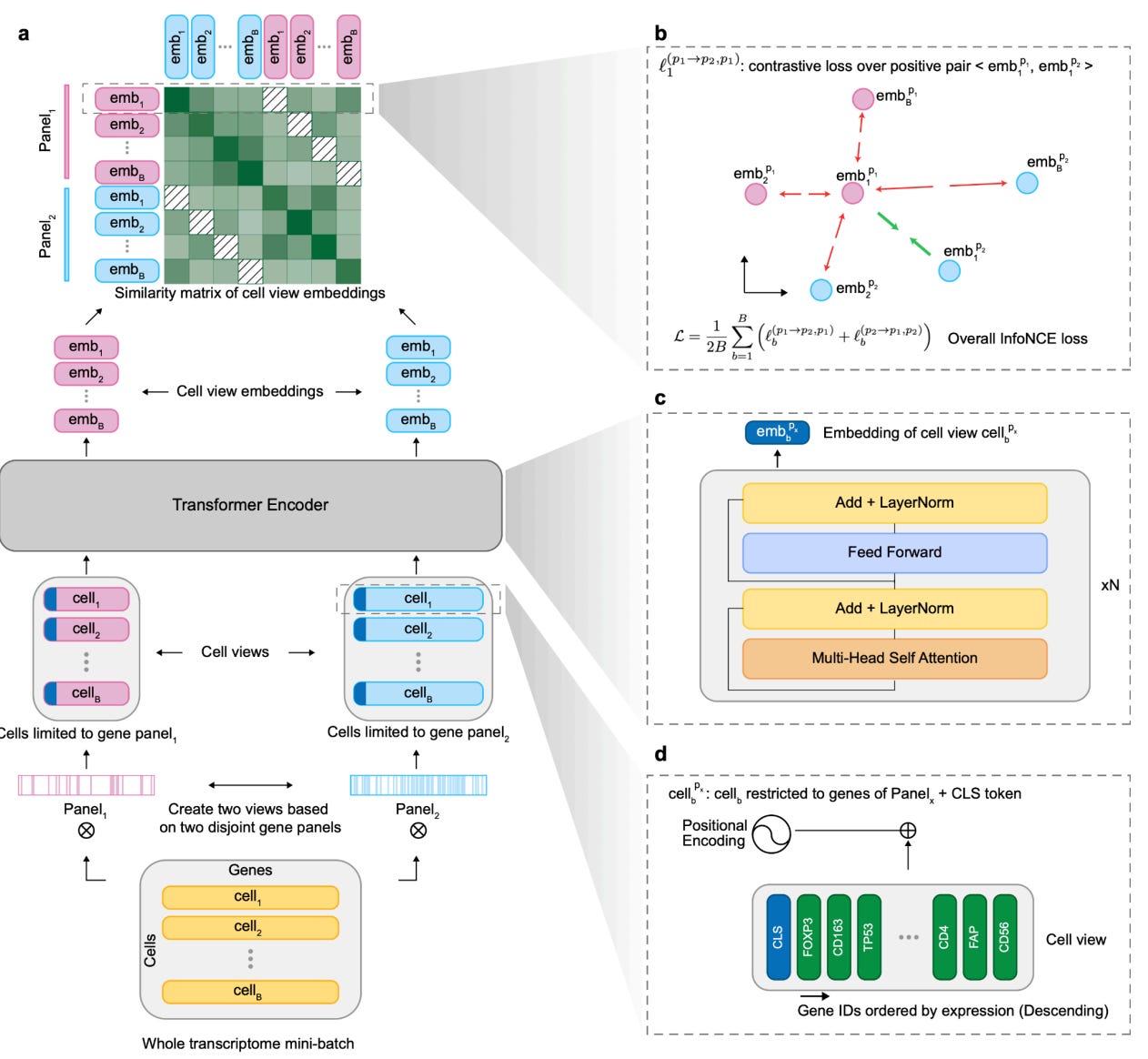
Technically, this is done using contrastive learning. Each cell is split into two disjoint gene subsets. The model sees both views and is trained to pull them together in embedding space, while pushing apart views from different cells.
To make this work, the team borrowed a trick from BERT: a dedicated CLS token. In language, CLS represents a sentence. In scConcept, it represents the entire cell. The loss is applied directly to that token.
“If the model can solve this task,” Mojtaba explained, “it has to develop a high-level idea of cell identity. There’s no shortcut.”
That framing turns the embedding from a side effect into the main objective. The model is no longer rewarded for local gene accuracy, but for capturing global cellular identity.
📊 Where the data work really shows
The architecture alone is only half the story. The other half lives in how the data is presented. One of the team’s biggest insights came from a failed experiment. Early versions of scConcept used the same binning strategies as other models. The result was large, technology-driven shifts in the embedding space.
“That was the moment we realised something was fundamentally wrong,” Mojtaba said. “The model was learning the technology, not the biology.”
The fix was surprisingly simple. Instead of feeding absolute expression values, scConcept uses rank encoding. Genes are ordered by expression within each cell. Only relative relationships matter.
If gene A is higher than gene B, the model sees that. The actual counts are ignored.
This turns out to be remarkably robust. Different assays may disagree on absolute numbers, but gene rankings tend to stay stable. Rank encoding strips away much of the batch effect before the model even starts learning.
Then comes gene subsetting. During training, the model constantly sees partial views of cells, including realistic gene panels from spatial technologies. This forces the embedding to stay stable even when most genes are missing.
The result is a representation that does not panic when faced with a 300-gene panel instead of a full transcriptome.
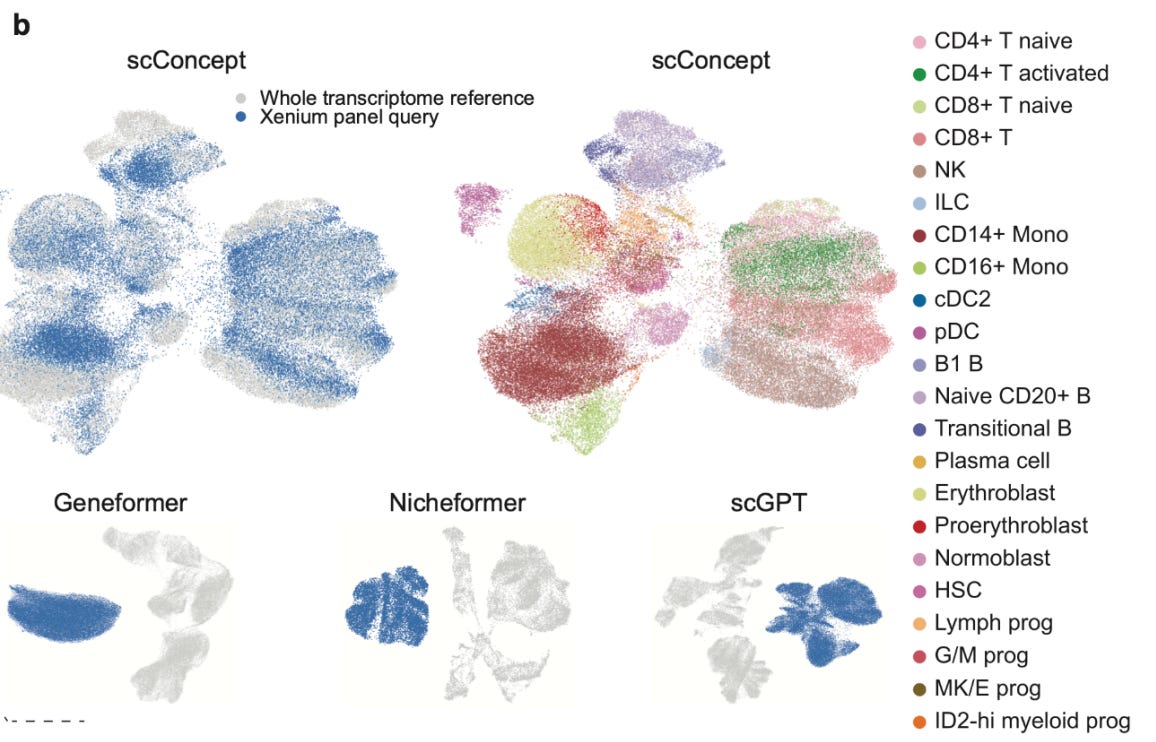
📢 Why it is different
The evaluation results reflect that design choice. scConcept consistently outperforms other foundation models on cell type annotation, cross-technology transfer, and spatial imputation, often matching or beating domain-specific tools. More importantly, it fails more gracefully.
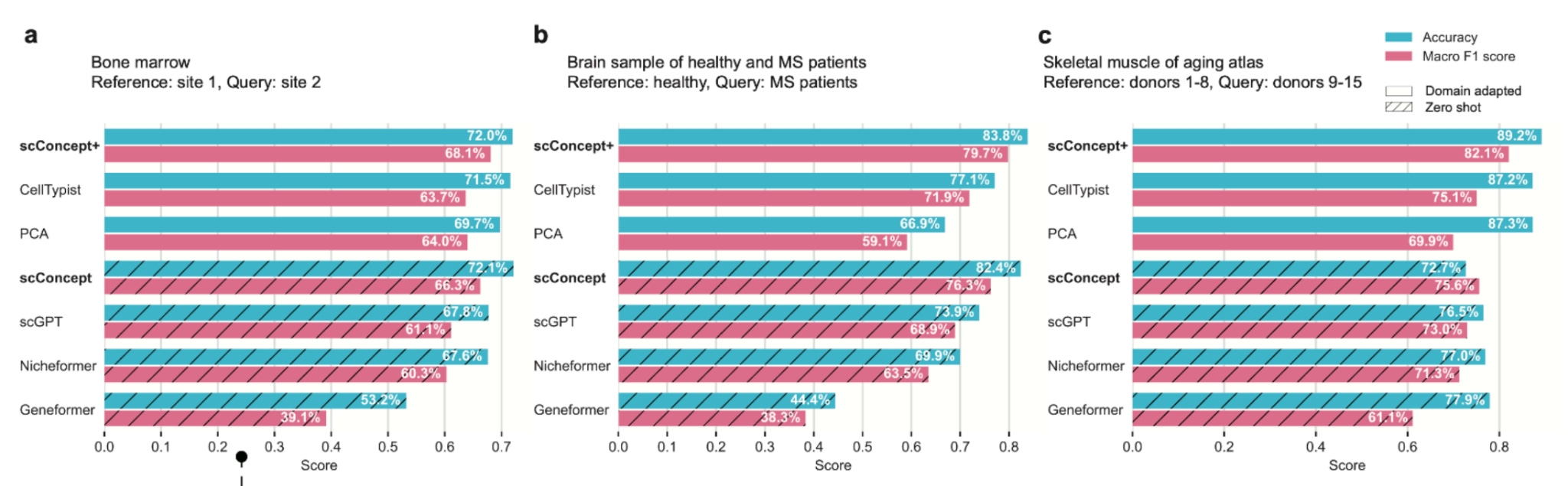
When information is missing, performance drops for the right reasons, not because the embedding space collapses. Closely related cell types remain close. Spatial structure is preserved. Adaptation improves things further without retraining the entire model.
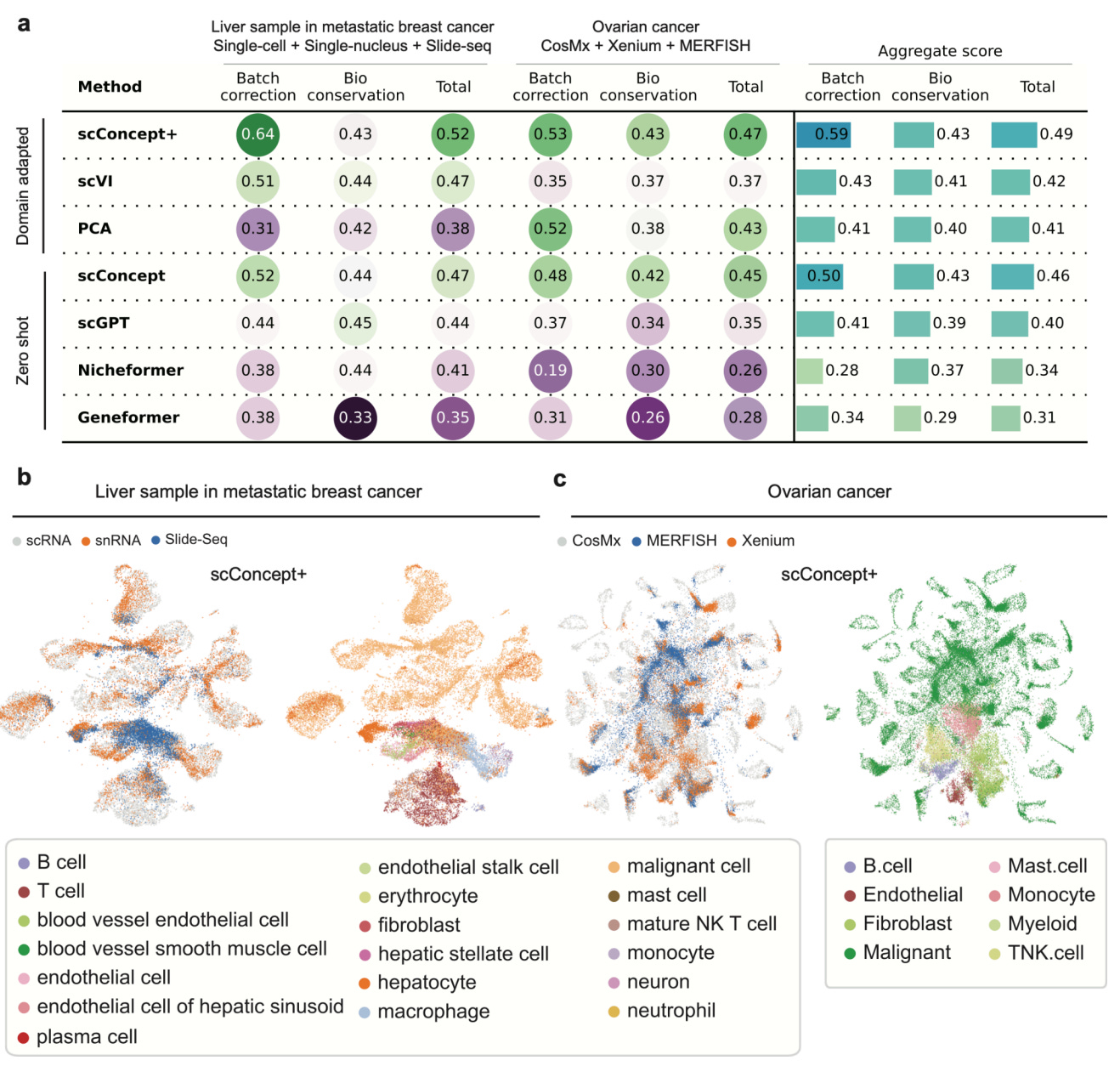
One subtle but important point the authors stress is that scConcept is not a batch correction method. It does not erase real biological differences. It just stops the model from confusing technology with biology.
That distinction matters if you actually want to interpret what the model is doing.
🔮 The Future
The long-term vision goes further than single cells. Right now, scConcept is trained on around 30 million cells, deliberately matching the scale of existing models. The next step is obvious: train on hundreds of millions. Initiatives like the Human Cell Atlas and repositories such as CELLxGENE now host hundreds of millions of publicly available cells, making this scale increasingly realistic.
However, the more interesting shift is conceptual. As Mojtaba put it, “Cells are the starting point. But biology doesn’t stop there. Tissues matter. Spatial context matters. Patients matter.”
Contrastive learning over partial views opens the door to representing tissue sections, neighbourhoods, even whole samples. Instead of asking whether two gene sets come from the same cell, future models might ask whether two regions come from the same tissue state.
That feels like a natural progression, especially as spatial assays become routine.
For now, scConcept is a reminder that better questions often beat bigger models. By focusing on what practitioners actually use, rather than what looks impressive on paper, it points toward a quieter but more useful future for single-cell AI, which is probably what the field needs right now.
👨🔬 Get in touch with Mojtaba.
📑 Read the paper.
💻 Check out the code.
Thanks for reading Kiin Bio Weekly!
💬 Get involved
We’re always looking to grow our community. If you’d like to get involved, contribute ideas or share something you’re building, fill out this form or reach out to me directly.
Subscribe now to stay at the forefront of AI in Life Science and keep up with this upcoming season of deep dives.
Connect With Us
Have questions on this or suggestions for our next deep dive? We’d love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website