
Seoul National University’s FoldMason, NIH’s RNAnneal, and Harvard’s ARK
Kiin Bio's Weekly Insights

Welcome back to your weekly dose of AI news for Life Science!
What’s your biggest time sink in the drug discovery process?
🧬 FoldMason: Structure Alignment at Database Scale
Can we align hundreds of thousands of protein structures without everything grinding to a halt?
Protein structure is far more conserved than sequence, which is why structural alignment is so powerful for studying distant evolutionary relationships. The problem has always been speed. Accurate multiple structure alignment tools exist, but they do not scale to the size of modern resources like AlphaFoldDB.
That is the problem FoldMason, from researchers at Seoul National University, is built to solve.
FoldMason is a progressive multiple protein structure alignment method based on the 3Di structural alphabet from Foldseek. By encoding local tertiary interactions as fast, sequence-like representations, it makes structure-level alignment feasible at a scale that was previously unrealistic.
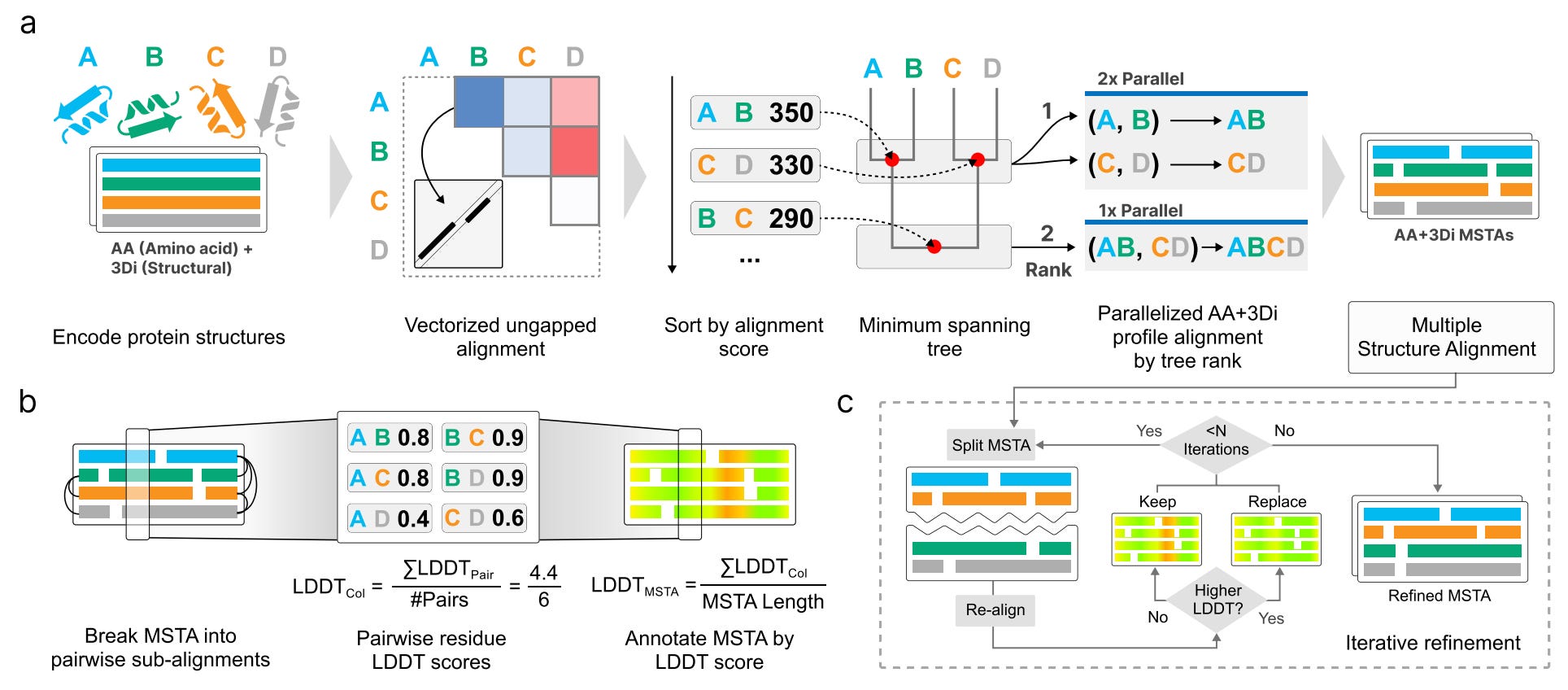
🔬 Applications and Insights
1️⃣ Structure alignment at near-sequence speeds
FoldMason achieves accuracy comparable to gold-standard aligners such as MUSTANG and mTM-align while running two to three orders of magnitude faster. On large AlphaFoldDB clusters, it delivers speedups of up to 700×.
2️⃣ Reference-free alignment quality scoring
Instead of relying on curated reference alignments, FoldMason extends the LDDT metric to score entire multiple structure alignments, providing a structure-aware quality signal that scales to very large datasets.
3️⃣ Handles flexible proteins better than superposition
For proteins such as calmodulin, globins, and odorant-binding proteins, FoldMason achieved up to 23 per cent higher LDDT than superposition-based methods that struggle with domain motion.
4️⃣ Structure-based phylogeny beyond the twilight zone
On Flaviviridae glycoproteins, FoldMason-derived phylogenies closely matched manually curated structural trees, achieving quartet similarity above 88 per cent with improved bootstrap support.
💡 Why It’s Cool
FoldMason turns structure alignment from a specialist bottleneck into a routine operation. In the post-AlphaFold era, the challenge is no longer predicting structures, but comparing them at scale. This is a big step in that direction.
📖 Read the paper
🧬 RNAnneal: Predicting RNA Structure Ensembles From Sequence
Can we predict the full set of RNA shapes from sequence alone?
RNA does far more than shuttle information. Its three-dimensional folds control splicing, regulation, catalysis, and viral replication. But RNA rarely sits still. It exists as an ensemble of structures, constantly shifting between states.
That is the challenge RNAnneal, from researchers at the NIH, Stony Brook University, and collaborators, sets out to tackle.
RNAnneal is an ab initio RNA structure ensemble prediction method that combines deep generative modelling with physics-inspired annealing. Instead of producing a single best fold, it generates a distribution of conformations that better reflects how RNA behaves in reality.
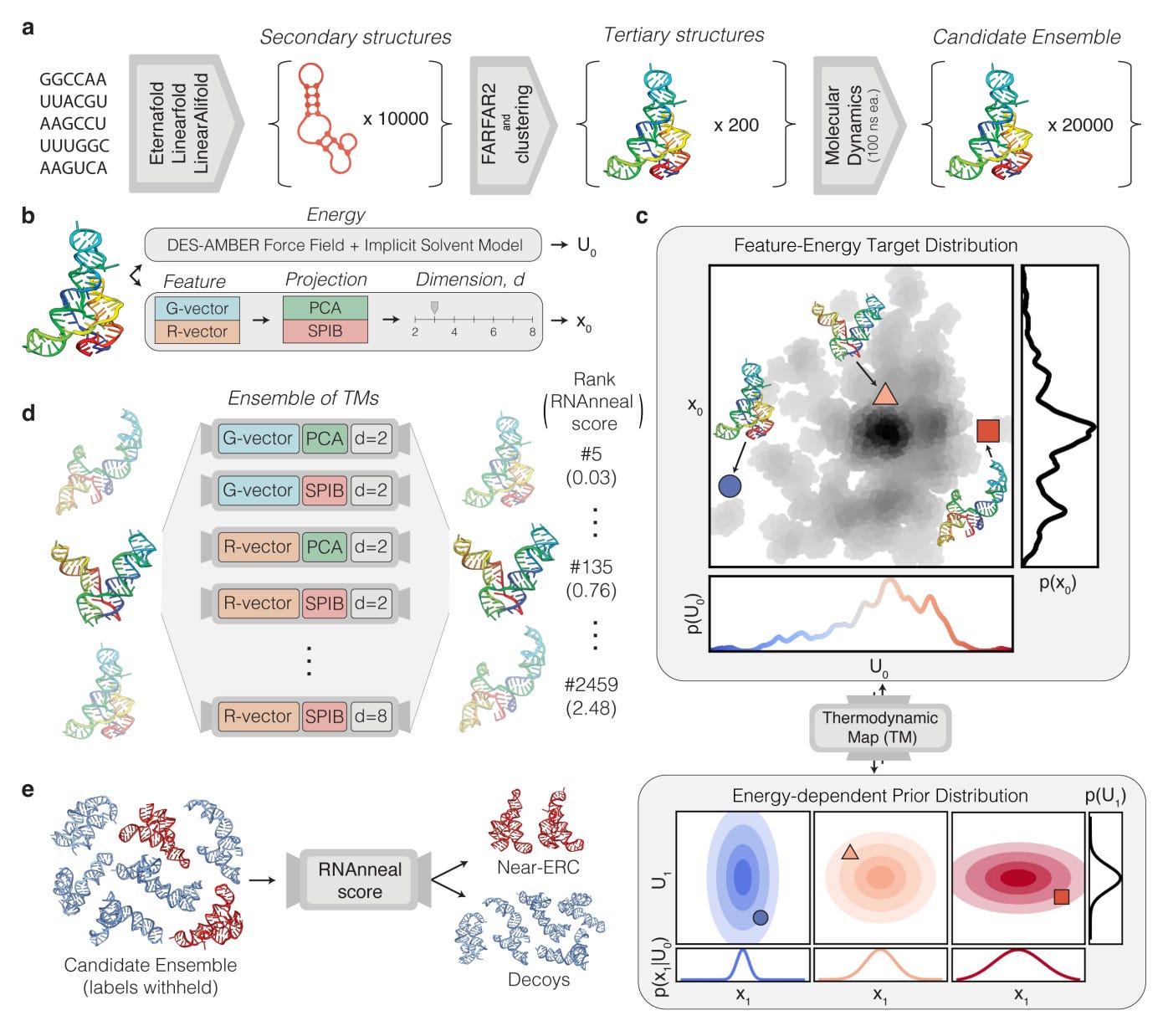
🔬 Applications and Insights
1️⃣ Ensemble rather than single-structure prediction
RNAnneal generates hundreds of candidate conformations per sequence and recovers experimentally observed ensemble populations, including low-probability but functionally important states.
2️⃣ Improved agreement with experimental data
Across benchmark RNAs, RNAnneal achieves up to two-fold higher correlation with SHAPE and NMR-derived ensemble distributions compared with single-structure predictors.
3️⃣ Physics-aware sampling improves realism
The annealing procedure reduces steric clashes and unphysical geometries, lowering average structural energy by 15–25 per cent relative to purely neural generative baselines.
4️⃣ Scales to longer and more complex RNAs
RNAnneal successfully models RNAs longer than 200 nucleotides, a regime where many ensemble methods either collapse to a single structure or fail.
💡 Why It’s Cool
RNAnneal shifts RNA structure prediction from snapshots to populations. For RNA therapeutics, ribozyme engineering, and antiviral design, knowing which shapes exist, and how often, matters just as much as knowing the lowest-energy fold.
📖 Read the preprint
💻 Code / platform: Not yet released
🧠 ARK: Adaptive Retrieval for Knowledge Graph Reasoning
Can language models explore knowledge graphs without getting lost or stuck?
Knowledge graphs are powerful, but querying them well is difficult. Some questions require broad coverage across entities. Others demand careful multi-hop reasoning. Most systems pick one strategy and hope it works.
That is the tension ARK, from researchers at Harvard Medical School, the Broad Institute, the University of Oxford, and collaborators, is designed to resolve.
ARK, short for Adaptive Retriever of Knowledge, is an agentic system that allows a language model to decide when to search broadly and when to explore locally. It uses just two tools: global lexical search over node text and one-hop neighbourhood exploration that composes into multi-hop reasoning.
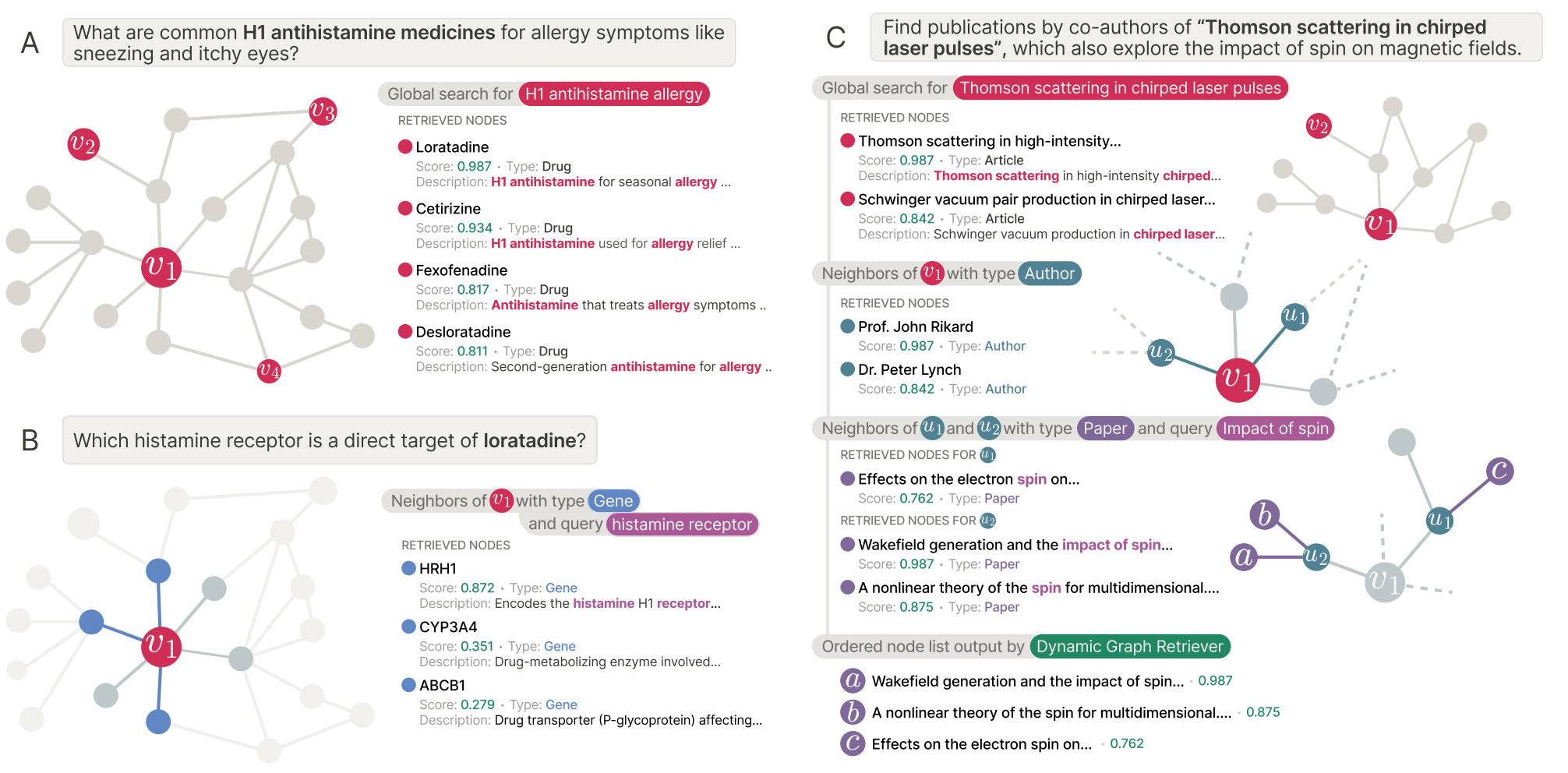
🔬 Applications and Insights
1️⃣ Strong gains without task-specific training
On the STaRK benchmark, ARK achieves 59.1 per cent Hit@1 and 67.4 MRR, improving Hit@1 by up to 31.4 percentage points over training-free and retrieval-only baselines.
2️⃣ Automatically adapts to query type
For text-heavy queries, ARK relies on global search nearly 88 per cent of the time. For relational queries, it shifts toward neighbourhood exploration, using it over half the time on MAG and PRIME.
3️⃣ Robust across very different graphs
ARK performs consistently across e-commerce, academic, and biomedical knowledge graphs. On PRIME, it achieves 48.2 per cent Hit@1, second only to reinforcement-learning-based approaches.
4️⃣ Distillation without labels actually works
By imitating tool-use trajectories, an 8B distilled model retains up to 98.5 per cent of the teacher model’s Hit@1 while improving performance by 26.6 points over the base model.
💡 Why It’s Cool
ARK treats retrieval as a decision process rather than a fixed pipeline. It balances global context with local reasoning in a way that mirrors how humans search. For graph-based RAG and scientific question answering, that balance matters.
📖 Read the paper
💻 Code / platform: Research prototype
Thanks for reading!
💬 Get involved
We’re always looking to grow our community. If you’d like to get involved, contribute ideas or share something you’re building, fill out this form or reach out to me directly.
Connect With Us
Have questions or suggestions? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website