
Shandong’s MetaMDA, University of Queensland’s Structome Toolkit, and CMU’s OMTRA
Kiin Bio's Weekly Insights

Welcome back to your weekly dose of AI news for life science.
What’s your biggest time sink in the drug discovery process?
🦠 MetaMDA: A New Way to Predict Microbe-Drug Interactions
What if we could predict how microbes interact with drugs and explain why those interactions occur?
Microbes can influence the absorption, distribution, metabolism and excretion of drugs. Drugs, in turn, can reshape microbial communities. Yet most computational methods fail to explain their predictions and cannot handle drugs or microbes missing from the training data.
MetaMDA, developed at Shandong University, takes a different approach. It builds a heterogeneous graph of microbes, metabolites and drugs, applies a random-walk model to capture relationships, and uses an XGBoost classifier to generate explainable microbe-drug predictions.
Because metabolites are included as explicit nodes, the model can propose mechanistic pathways that link microbes and drugs through shared biochemical intermediates.
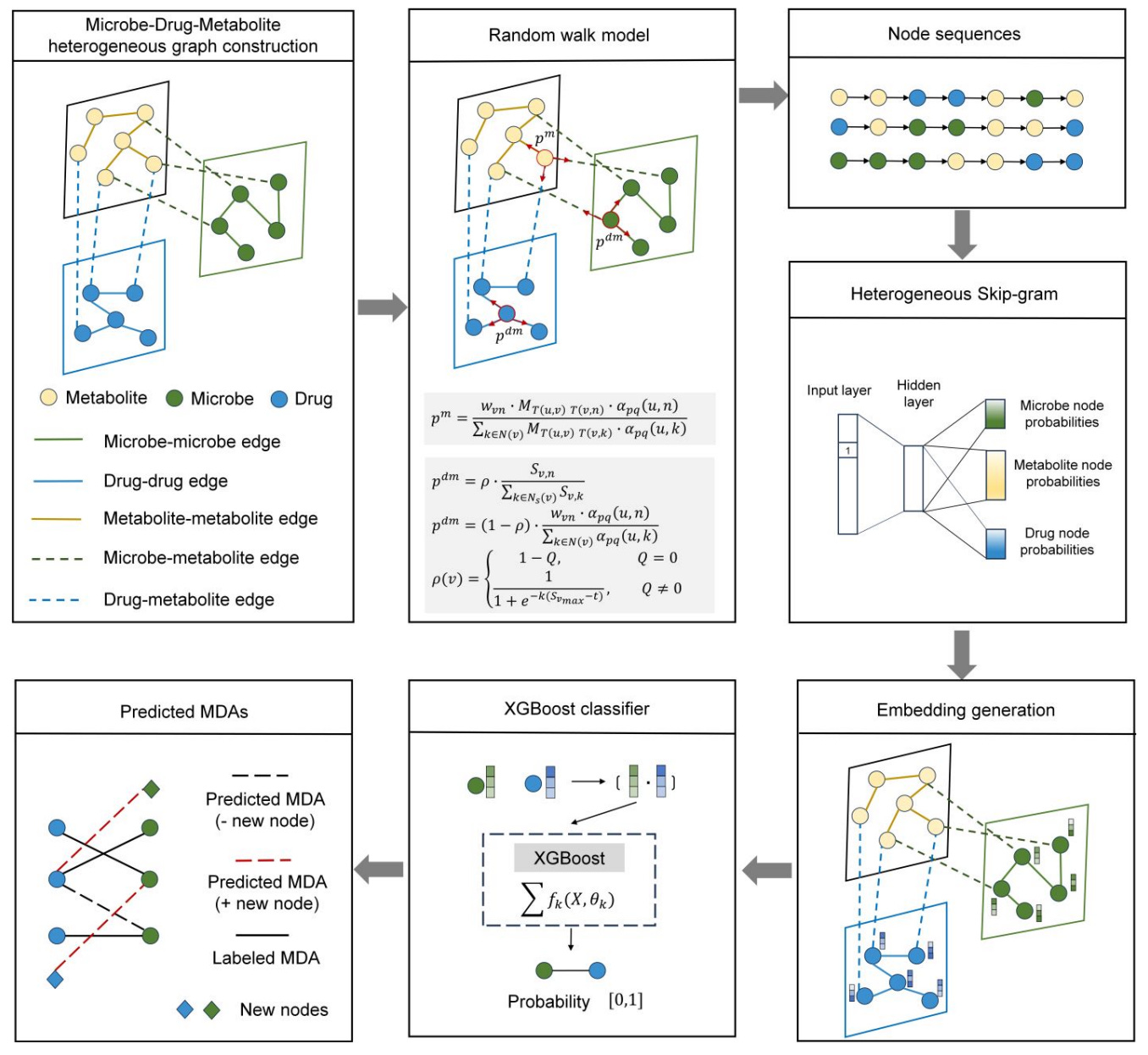
🔬 Applications and Insights
1️⃣ Stronger predictive performance
MetaMDA outperformed current methods by an average of 26 per cent across multiple benchmark datasets.
2️⃣ Validated antibiotic associations
For tetracycline and vancomycin, all top 10 predicted microbe–drug interactions matched previously published findings, demonstrating strong real-world grounding.
3️⃣ Predicts associations for unseen drugs and microbes
MetaMDA successfully predicted interactions involving microbes or drugs absent from the training set, such as microbe–acarbose links that were confirmed in external studies at an 85 per cent rate.
4️⃣ Biological explanations through metabolites
By tracing predictions through metabolite nodes, MetaMDA can propose hypotheses such as an E. coli interaction with escitalopram mediated through glutamate.
💡 Why It Is Cool
MetaMDA helps uncover how microbiomes influence drug response and highlights pathways that could support more personalised therapies. It is a step toward models that do not only predict interactions but also explain them biologically.
📄 Read the paper
⚙️ Try the code.
Big thanks to our newest contributor, Yaa, for writing this week’s article on MetaMDA. Great find! We’re looking forward to sharing more of your insights.
If you’re interested in contributing or joining the community, reach out to me or fill this out!
🧬 Structome-TM and Structome-AlignViewer: Structure-First Phylogenomics
What if we could recover evolutionary relationships even when sequences have diverged beyond recognition?
Sequence-based phylogenomics is powerful, but once proteins diverge deeply, the signal collapses and entire families become invisible to traditional alignment. Structure, however, is far more conserved than sequence and can reveal distant homology that sequence-only tools miss.
Researchers led by Ashar Malik introduce two complementary tools that bring structure into evolutionary analysis: Structome-TM and Structome-AlignViewer.
Structome-TM uses TM-score to detect local fold similarity even when proteins differ significantly in size. Structome-AlignViewer layers per-column confidence onto 3D models, highlighting which structural regions are truly conserved.
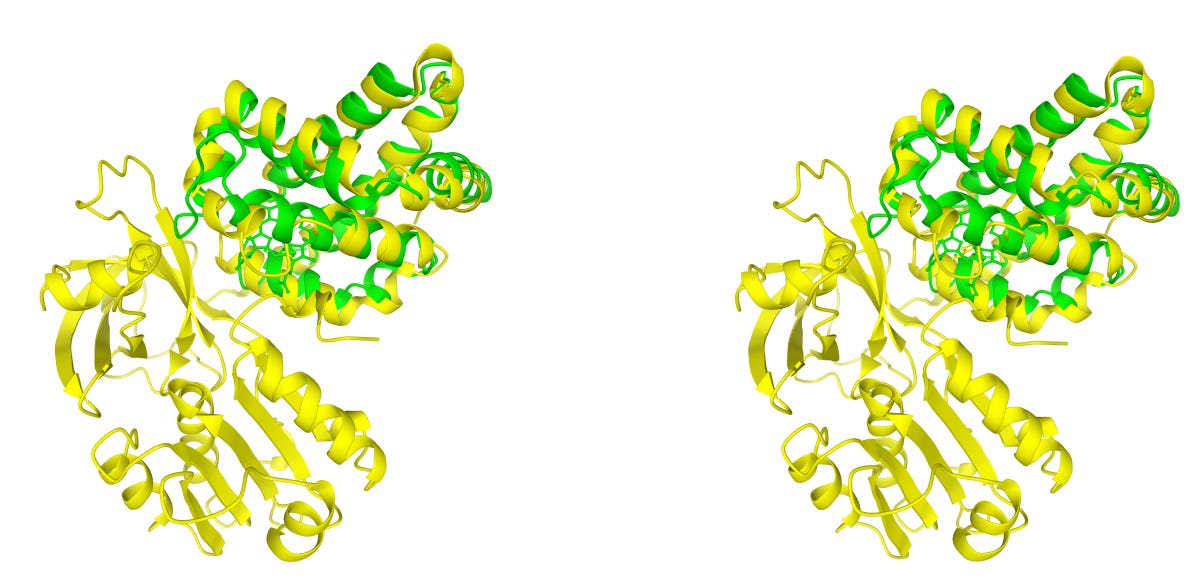
🔬 Applications and Insights
1️⃣ Deep homology detection
Structome-TM identifies homologues that global metrics overlook. A globin domain that scores only 0.213 under a sequence-based global alignment shows a TM-score of 0.803, correctly revealing its evolutionary relationship.
2️⃣ Confidence-aware structural alignments
Structome-AlignViewer benchmarks per-column confidence against thousands of SCOP and CATH alignments, making conserved structural cores easy to identify and gap-rich regions easy to trim.
3️⃣ Cleaner, more reliable phylogenies
By restricting analysis to structurally validated positions, users build evolutionary trees that remain stable even when sequence identity is extremely low.
4️⃣ Functional insight beyond sequence
Structural conservation highlights residues essential for stability and function, supporting:
• rational protein engineering
• prediction of pathogen escape mutations
• understanding drug-resistance mechanisms
• annotation of dark-proteome proteins with no detectable sequence homologues
💡 Why It Is Cool
Structural phylogenomics goes beyond better trees. It turns 3D evolutionary patterns into tools for protein engineering, pathogen surveillance and drug discovery, especially when sequence information is too weak to rely on.
📄 Read the papers: StructomeTM and Structome-AlignViewer
⚙️ Try the codes: Structome-TM and Structome-AlignViewer
⚛️ OMTRA: One Model for All Structure-Based Drug Design Tasks
What if one AI system could design molecules, generate conformers and perform docking, all within the same architecture?
Structure-based drug design currently relies on specialised models that do not transfer across tasks. Molecule generators cannot dock, docking models cannot generate conformers, and pharmacophore tools cannot design new ligands.
Researchers at Carnegie Mellon and Pittsburgh introduce OMTRA, a unified generative framework that treats ligands, protein pockets and pharmacophores as multimodal graphs.
Its multimodal flow-matching architecture supports flexible conditioning and is pretrained on 500 million 3D conformers, enabling a single model to solve multiple SBDD tasks.
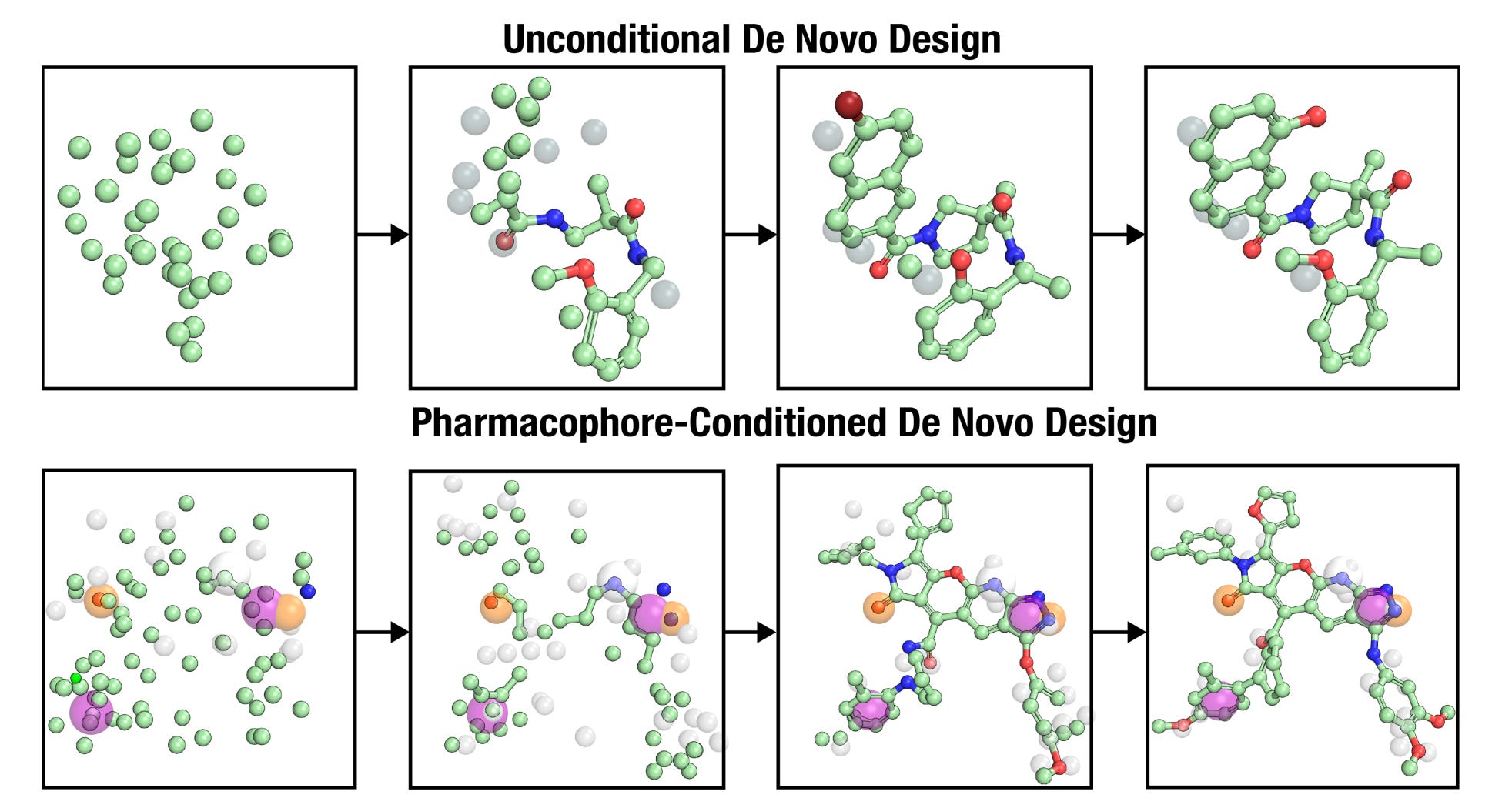
🔬 Key Results and Applications
1️⃣ Pocket-conditioned de novo design
OMTRA reaches around 90 per cent PoseBusters validity and produces ligands with native-like interactions 37 per cent more often than the next best method.
2️⃣ State-of-the-art redocking
On PoseBusters, OMTRA achieves 92 per cent top-1 RMSD ≤ 2 Å, outperforming SurfDock, Gnina and even AlphaFold3 when pocket residues are provided.
3️⃣ Pharmacophore-guided molecule generation
Pharmacophore conditioning improves interaction recovery from approximately 51 per cent to 61 per cent. OMTRA achieves more than 97 per cent pharmacophore matching and improves docking accuracy to 99 per cent RMSD ≤ 2 Å in top-5 predictions.
4️⃣ Insights into multi-task learning
Large-scale ligand-only pretraining and multi-task training yielded modest improvements, suggesting that true cross-task transfer remains a challenging frontier.
💡 Why It Is Cool
OMTRA shows that SBDD tasks do not need separate models. One architecture can handle ligand generation, docking and pharmacophore conditioning, paving the way for more unified and interpretable AI tools in drug discovery.
📄 Read the paper.
⚙️ Try the code
Thanks for reading Kiin Bio Weekly!
💬 Get involved
We’re always looking to grow our community. If you’d like to get involved, contribute ideas or share something you’re building, fill out this form or reach out to me directly.
Connect With Us
Have any questions or suggestions for a post? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website