
Stanford's GATSBI, ProtiCell and MIT's StriMap
Kiin Bio's Weekly Insights

Welcome back to your weekly dose of AI news for Life Science!
Keeping up with AI x life science news can get exhausting.
It’s scattered across LinkedIn, X, Substack, arXiv, Slack, newsletters... and you still somehow miss the things that actually matter. Too much noise, not enough signal.
We’re building something to fix that: a smarter, more powerful way to stay on top of what’s actually relevant to you.
But we want to build it with you, not just for you. Take 2 minutes to tell us what’s missing. What you share will directly shape what we build, and you’ll be the first to benefit from it.
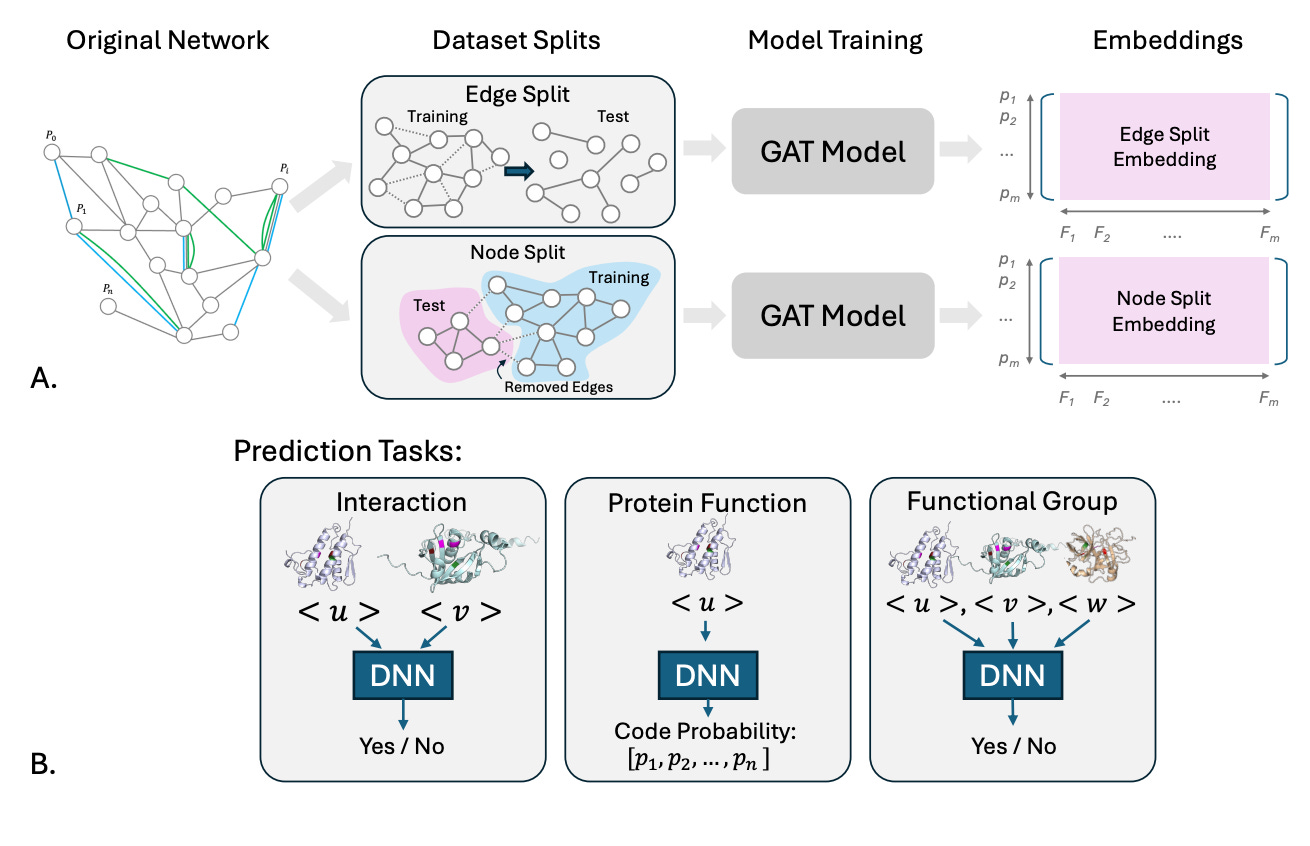
GATSBI: Improving Context-Aware Protein Embeddings Through Biologically Motivated Data Splits
🔬 Protein embeddings power everything from interaction prediction to functional annotation. But how we evaluate them matters just as much as how we build them. Random train/test splits let models cheat by memorizing well-connected proteins, inflating performance on benchmarks while hiding failures on the long tail of understudied proteins.
GATSBI (Graph Attention with Split-Boosted Inference) is a framework from Stanford that builds context-aware protein embeddings by integrating heterogeneous biological data, including PPIs from STRING, co-expression patterns, tissue-specific functional associations from HumanBase, and ESM-2 sequence representations, into a unified graph attention network.
🧬 The network covers 18,049 human proteins with 1.5M+ edges across three relationship types. Graph attention layers learn to weight different edge types and tissue contexts during message passing, producing embeddings that capture both local interactions and broader functional context.
⚡ The key insight is evaluation design. GATSBI introduces biologically motivated splits: edge splits (testing unseen relationships between known proteins) and node splits (testing entirely unseen proteins with less than 30% sequence identity). Across interaction prediction, function classification, and functional set prediction, GATSBI consistently outperforms PINNACLE, with the largest gains for understudied proteins (AUROC +0.259, AUPRC +0.290 on functional sets).
🔬 Applications and Insights
1️⃣ Plug-and-Play Protein Representations
Embeddings are pretrained and downloadable, making them drop-in features for downstream models predicting interactions, function, or pathway membership without retraining the graph.
2️⃣ Honest Benchmarking for Understudied Proteins
The node split reveals how models actually perform on proteins with limited prior evidence, exposing a critical gap that random splits conceal in current benchmarking.
3️⃣ Biologically Plausible “False Positives”
High-confidence predictions flagged as errors turned out to reflect real but unannotated relationships, suggesting the model captures genuine biology beyond current annotations.
4️⃣ Boosting the Long Tail of the Proteome
The heterogeneous graph adds the most information for low-degree proteins, precisely the understudied targets where computational predictions matter most for discovery.
💡 Why This Is Cool
The lesson here goes beyond protein embeddings. How you split your data determines what your benchmark actually measures. GATSBI shows that when you evaluate properly, the gap between methods changes dramatically, and models that look equivalent on well-studied proteins diverge sharply on the understudied ones that matter for real discovery.
📄 Read the paper.
💻 Try the code.
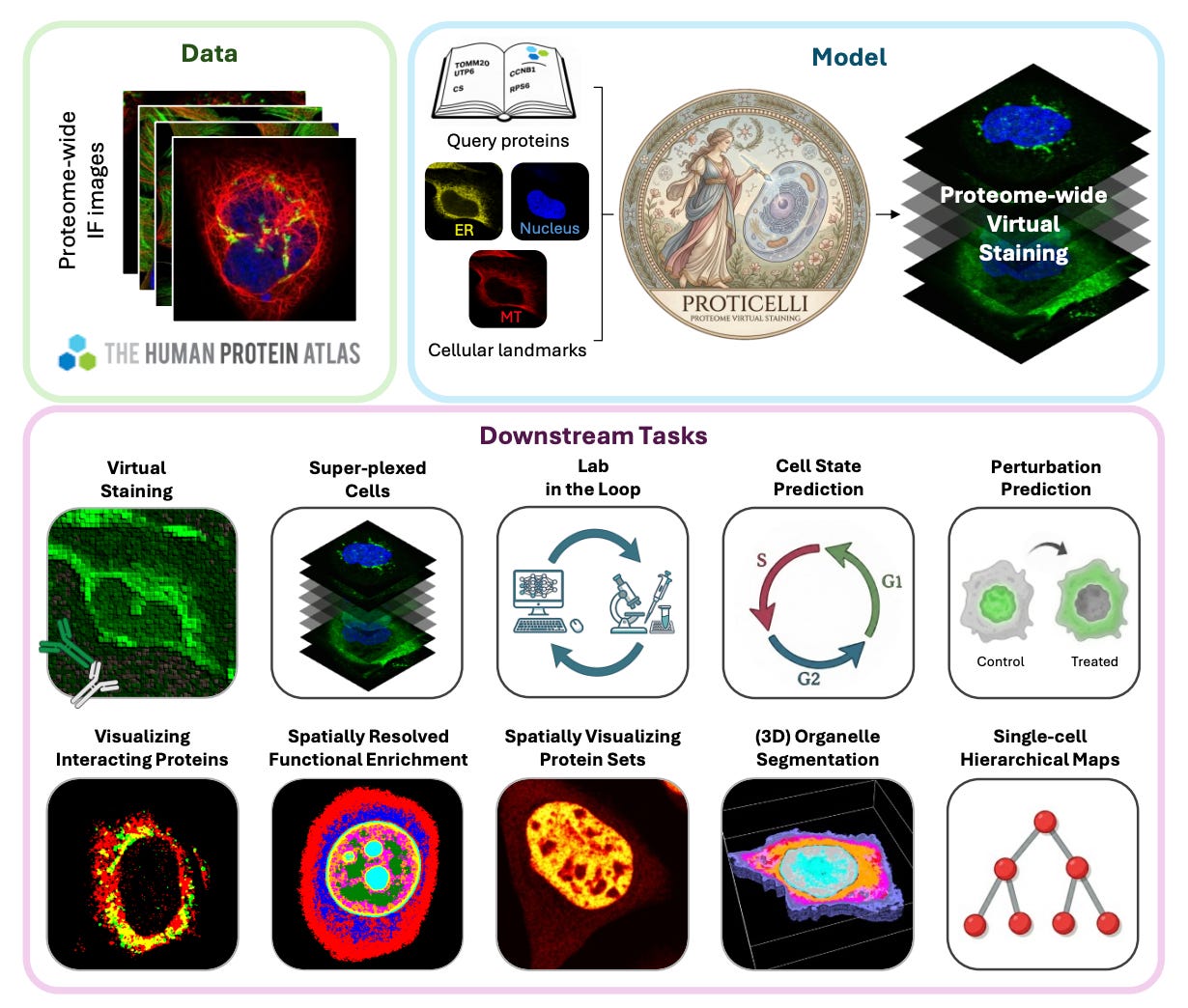
ProtiCelli: Generative Machine Learning Unlocks the First Proteome-Wide Image of Human Cells
🔬 Current imaging technologies can visualize tens of proteins simultaneously. A single human cell contains thousands. This gap means we still lack a complete picture of how the proteome is spatially organized, and brute-force experimental coverage is not practical at this scale.
ProtiCelli is a deep generative model from Stanford and KTH, trained on 1.23 million immunofluorescence images from the Human Protein Atlas, that simulates microscopy images for 12,800 human proteins using just three cellular landmark stains as input (nucleus, microtubules, ER).
🧬 Given only the landmark channels, the model predicts what the protein channel would look like, effectively hallucinating biologically accurate fluorescence patterns. It generalizes to unseen cell types and drug perturbations, preserves hierarchical subcellular organization, and even recapitulates known protein-protein interactions from the spatial patterns alone.
⚡ The team generated Proteome2Cell: 30.7 million simulated images representing 2,400 virtual cells across 12 human cell lines, now integrated into the Human Protein Atlas. The model can also infer drug-induced changes in protein localization from cell morphology alone, without ever seeing the drug-treated protein images during training.
🔬 Applications and Insights
1️⃣ Virtual Proteome-Scale Experiments
Enables imaging experiments that would take years and millions of dollars to run physically, compressing exploration of protein spatial organization into a single model inference.
2️⃣ Drug Mechanism-of-Action Discovery
Predicts how compounds alter protein localization from cell morphology alone, without staining for every target, potentially accelerating early-stage perturbation screening.
3️⃣ Orthogonal Interaction Signal
Predicted co-localization patterns correlate with known protein-protein interactions, providing a spatial signal that complements sequence-based interaction predictions.
4️⃣ A Public Resource for the Community
Proteome2Cell (30.7M images across 12 cell lines) is freely available through the Human Protein Atlas, giving researchers a shared baseline for cell biology and drug discovery.
💡 Why This Is Cool
This is the microscopy equivalent of protein structure prediction. Just as AlphaFold gave us predicted structures for proteins we had not crystallized, ProtiCelli gives us predicted images for proteins we have not stained. The fact that drug-induced changes emerge from morphology alone suggests the model has learned something genuinely deep about the relationship between cell shape and protein organisation.
📄 Read the paper.
💻 Try the code.
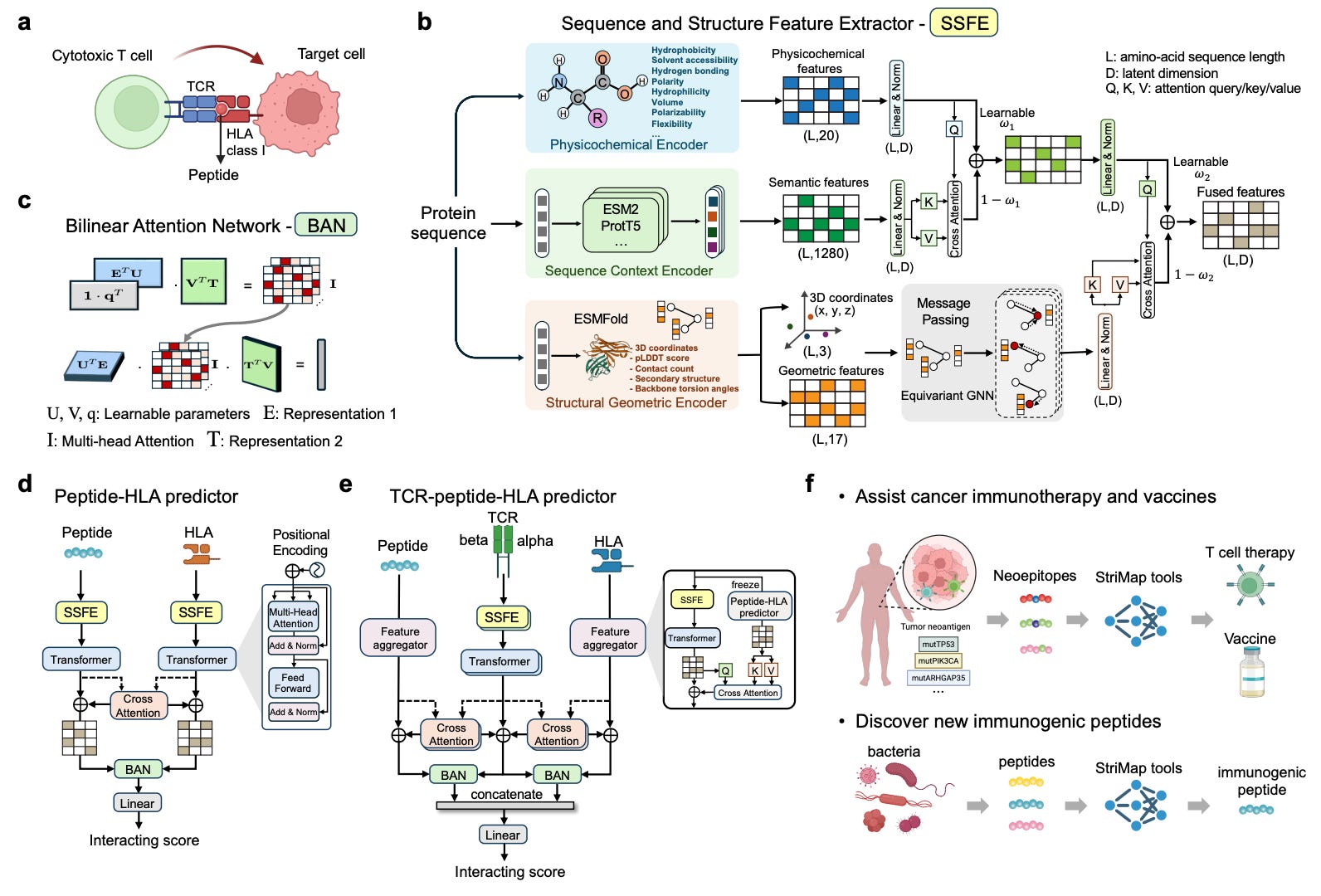
StriMap: A Structure-Informed Deep Learning Framework for TCR-Peptide-HLA Interactions
🔬 T cell receptor (TCR) recognition of peptide-HLA complexes drives adaptive immunity, from fighting infections to rejecting tumors to triggering autoimmunity. But predicting which TCR will bind which peptide-HLA is notoriously difficult: the interaction interface is flexible, the sequence space is vast, and training data is sparse.
StriMap is a unified deep learning framework from the Xavier lab at the Broad Institute and MIT that integrates physicochemical features, sequence context, and structural information at the recognition interface to predict TCR-peptide-HLA binding.
🧬 Rather than relying on sequence alone, StriMap models the 3D contact geometry between TCR CDR loops and the peptide-HLA surface. By combining structural priors with learned sequence representations, it captures binding patterns that pure sequence models miss, achieving state-of-the-art performance with improved generalizability across diverse datasets.
⚡ The real validation came from application: the team screened 13 million peptides from 43,241 bacterial proteins to find molecular mimics relevant to ankylosing spondylitis (AS). Top candidates were experimentally validated to activate T cells expressing an AS-associated TCR, and one peptide showed enrichment in inflammatory bowel disease patients, suggesting shared microbial triggers between AS and IBD.
🔬 Applications and Insights
1️⃣ Generalizable TCR Binding Prediction
Structure-informed features at the binding interface improve generalization beyond the peptides and HLAs seen during training, breaking a critical bottleneck for TCR prediction models.
2️⃣ Computational-to-Experimental Discovery Pipeline
The AS screening demonstrates a complete workflow: computational prediction of 13M peptides narrowed to candidates that were experimentally validated to activate disease-relevant T cells.
3️⃣ Cross-Disease Molecular Mimicry
A top validated microbial peptide showed enrichment in IBD patients, supporting the hypothesis that shared bacterial triggers drive immune cross-reactivity between AS and IBD.
4️⃣ Dual Application in Cancer and Autoimmunity
The framework applies to both cancer neoantigen prediction and autoimmune antigen discovery, bridging two major areas of immunotherapy.
💡 Why This Is Cool
TCR-antigen prediction has been one of the stubbornest problems in computational immunology. Most models fail to generalize beyond their training peptides. By bringing structural information into the prediction, StriMap breaks out of that limitation, and the AS/IBD discovery shows this is not just a benchmark improvement but a tool that finds real biology.
📄 Read the paper
💻 Try the code.
🗓️ Events & Competitions
The best competitions, hackathons, and community challenges in AI x life sciences, curated weekly. Know something worth featuring? Reply and let us know.
More upcoming events:
Agentic Genomics: Hands-on with AI for Variant Interpretation and GWAS | April 22, Virtual
A free, two-session workshop co-hosted by Manuel Corpas and Segun Fatumo, running entirely in Google Colab. No setup, no cost, just a browser. Session one covers variant interpretation using Ensembl VEP, ClinVar, and ACMG criteria. Session two runs a full GWAS pipeline with ClawBio, including polygenic risk scores and locus fine-mapping. Built to remove barriers for researchers anywhere in the world.
Register (free): https://luma.com/vc98zeik
BioHackathon Europe 2026 | November 9-13, Barcelona
ELIXIR’s annual international bioinformatics hackathon, running since 2018. 160+ participants, five days of collaborative coding on open bioinformatics infrastructure and tools. The call for project proposals opens March 16 and closes April 15 - so if you want to lead a project, that’s your window.
Thanks for reading!
💬 Get involved
We’re always looking to grow our community. If you’d like to get involved, contribute ideas or share something you’re building, fill out this form or reach out to me directly.
Connect With Us
Have questions or suggestions? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website