
Stanford's Proteo-R1, Hamburg's ActivityFinder, and NUS's ProteinConformers
Kiin Bio's Weekly Insights

Welcome back to your weekly dose of AI news for Life Science!
We know how hard science is. That’s why we built the Pioneer Programme.
We’re selecting academic and nonprofit research teams to get one year of free access to our drug discovery platform plus hands-on support from our science team. If your research bottleneck isn’t data but connecting the findings you already have, this is for you.
We’re looking for teams asking: which targets should we prioritise? How do we interpret conflicting evidence across datasets? Which hypotheses are worth testing next? Where are the strongest translational opportunities?
No cost. No data transfer. All IP stays with your institution. Applications close August, cohort starts September.

Proteo-R1: Reasoning Foundation Models for De Novo Protein Design
🔬 Current deep learning methods for protein design generate molecular structures without explicitly reasoning about which residues matter or why. All residues are treated uniformly during generation, and design intent is implicitly buried in diffusion parameters. This makes it hard to interpret why a model made a particular choice, or to reuse that logic on a different target.
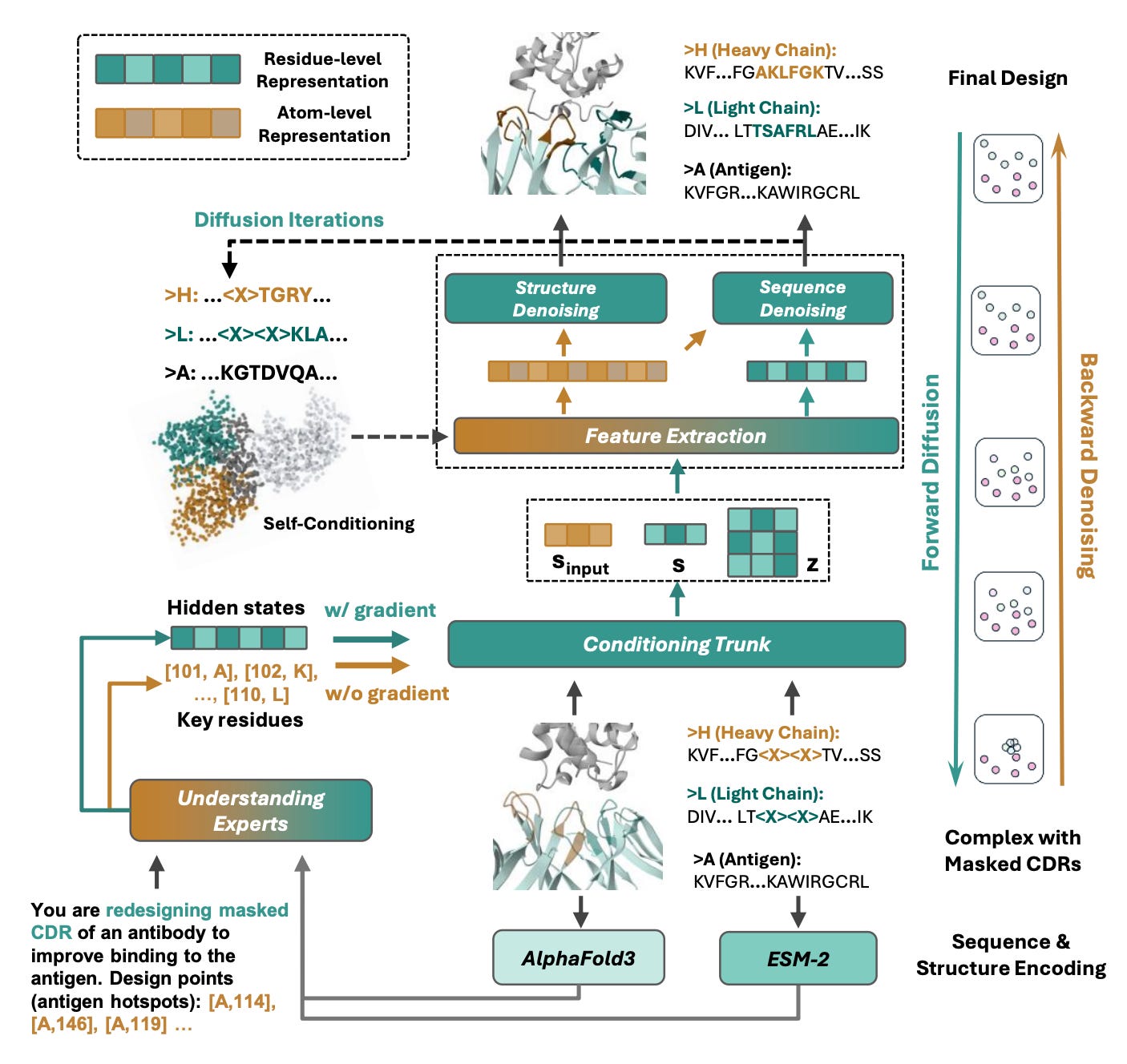
Researchers from Stanford, RIKEN, and collaborating institutions introduce Proteo-R1, a framework that separates molecular understanding from geometric generation. A multimodal large language model first reasons about binding interactions, then passes residue-level constraints to a diffusion model that generates the structure.
🧬 The understanding expert (a multimodal LLM) analyses protein sequences, AF3-style structural representations, and textual context to identify key interaction residues and predict their amino acid identities. These sparse, residue-level decisions are passed as hard constraints to the generation expert, an AlphaFold3-style diffusion model that performs conditional co-design while respecting the fixed interaction anchors. Training proceeds through three stages: multimodal alignment, structural reasoning mid-training, and joint reasoning-guided design on antibody-antigen complexes from SAbDab.
⚡ On simultaneous multi-CDR antibody redesign, Proteo-R1 achieves the lowest or near-lowest per-CDR RMSD in five of six regions, with interface improvement (IMP) of 56.58%. It produces the lowest steric clash rates (0.50% intra-chain, 0.14% inter-chain) and best dihedral distribution divergence among all tested methods. On CDR-H3 design via the RAbD benchmark, it obtains the best lDDT (0.9693), TM-score (0.9816), and DockQ (0.801) over DGENet, BoltzGen, and MFDesign.
🔬 Applications and Insights
1️⃣ Interpretable Antibody Engineering
The reasoning expert produces explicit, human-readable justifications for which residues it selects as interaction anchors. You can inspect and modify the design logic directly.
2️⃣ Controllable CDR Design
Researchers can edit the reasoning outputs (e.g. specifying different hotspot residues) to steer the generative model without retraining, giving fine-grained control over binding specificity.
3️⃣ Structure-Sequence Consistency
Proteo-R1 is the only method that achieves positive structure-sequence consistency (Δ = IF-AAR minus AAR) across five of six CDR regions. Its designs are structurally grounded rather than simply recovering native sequences.
4️⃣ Generative Backend Flexibility
The reasoning expert works with alternative generative frameworks. Tested with UniMoMo, it improves IMP from 65% to 67.79% and binding energy from 8.46 to 7.35 ΔG without architectural changes to either component.
💡 Why This Is Cool Proteo-R1 separates the “what should we build” question from the “how do we build it” question. That mirrors how human protein engineers actually work: identify critical interaction residues first, then optimise geometry under those constraints. The reasoning module plugs into different generative backends, and the design logic is readable and editable rather than locked inside a diffusion trajectory.
📄 Read the paper
💻 Try the code
ActivityFinder: Toward the Fully Automatic Integration of Structural and Binding Affinity Data
🔬 Building computational models that predict binding affinity requires both 3D protein-ligand structures and experimental activity measurements. These data live in separate databases (PDB for structures, ChEMBL for bioactivities) with no fully automated way to link them. Identifier-based approaches miss connections where sequences diverge, ligand representations vary, or binding-site mutations are present.
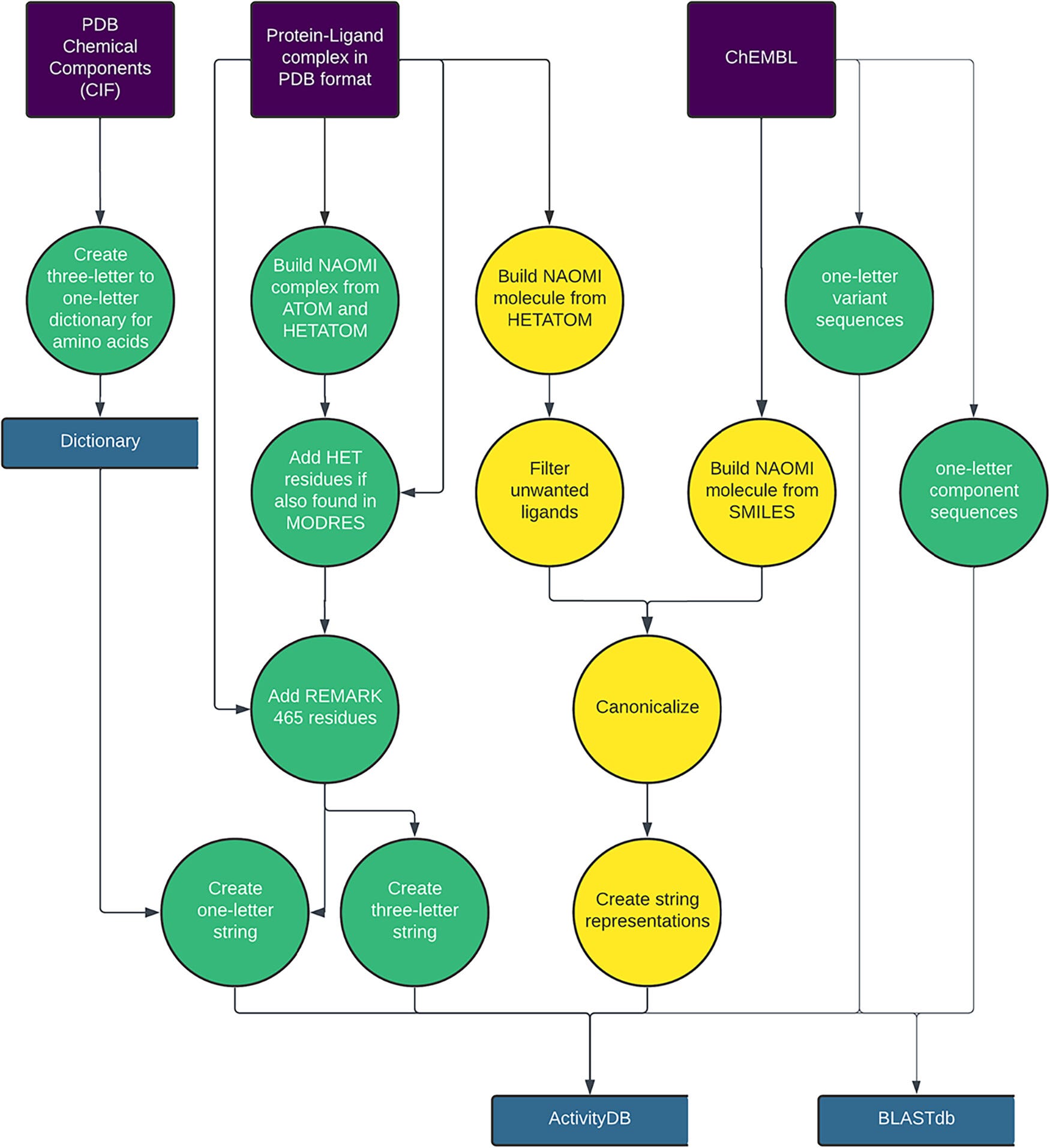
A team at the University of Hamburg developed ActivityFinder, a fully automated pipeline that links crystal structures of protein-ligand complexes directly to wet-lab bioactivity data. It requires only PDB files and a ChEMBL database dump, with no external services or continuous data connections.
🧬 ActivityFinder works in two stages. First, it builds an ActivityDB instance by parsing PDB structures into the NAOMI data format, extracting ligand representations as six different string types (InChI, InChIKey, canonical SMILES with and without stereo, InChI connection layer, InChI hydrogen layer), and creating BLAST databases from protein sequences. Second, it queries this database using sequence alignments (at 80%+ identity) and detailed chemical structure matching to cross-reference structural and bioactivity records. It tracks mutations and binding-site residues at atomic resolution.
⚡ Applied to 226,302 PDB structures and ChEMBL 35, ActivityFinder linked 20,197 PDB entries involving 13,734 PDB ligands to 17,829 unique ChEMBL ligands across 2,585 ChEMBL targets, covering over one million bioactivity data points. Compared to BioChemGraph (an identifier-based method), ActivityFinder identifies 46,287 unique structure-activity triplets versus 19,333. Of the 6,575 triplets unique to ActivityFinder, 1,082 are entirely novel: new combinations of PDB complex, ChEMBL target, and ChEMBL molecule.
🔬 Applications and Insights
1️⃣ Proprietary Data Integration
Because it requires only local PDB files and an SQL database, ActivityFinder works for pharmaceutical companies that want to link in-house crystal structures to internal bioactivity records without sending data to external services.
2️⃣ Mutation-Aware Linking
The tool explicitly tracks sequence variants and binding-site mutations at atomic resolution. Researchers can study how specific point mutations affect ligand binding affinity across related structures.
3️⃣ Training Data for ML Scoring Functions
The linked structure-activity pairs are ready-made training datasets for machine learning scoring functions. Quality annotations (confidence levels 1 to 5) let modellers filter for the precision their application requires.
4️⃣ Expanding Known Chemical Space
Of the novel triplets ActivityFinder uniquely identifies, many involve new combinations of PDB complex, ChEMBL target, and ChEMBL molecule not found by any other method. These are new data points for structure-based drug design.
💡 Why This Is Cool
Everyone building a scoring function or binding predictor eventually has to manually assemble their own structure-activity dataset. The results are rarely shared and identifier matching misses a lot. ActivityFinder automates this from scratch, works on proprietary data, and finds 2.4x more structure-activity links than the identifier-based alternative.
📄 Read the paper
💻 Try the tool: Available via the ProteinsPlus REST API
ProteinConformers: Large-Scale and Energetically Profiled Descriptions of Protein Conformational Landscapes
🔬 Understanding protein function requires capturing how structures move across their conformational space. Existing MD trajectory databases start only from native structures (near the global energy minimum), conformer generators have no standardised benchmarks, and available datasets provide limited energetic annotations. No resource maps the full spectrum from non-native to near-native states with both structural and energetic characterisation.
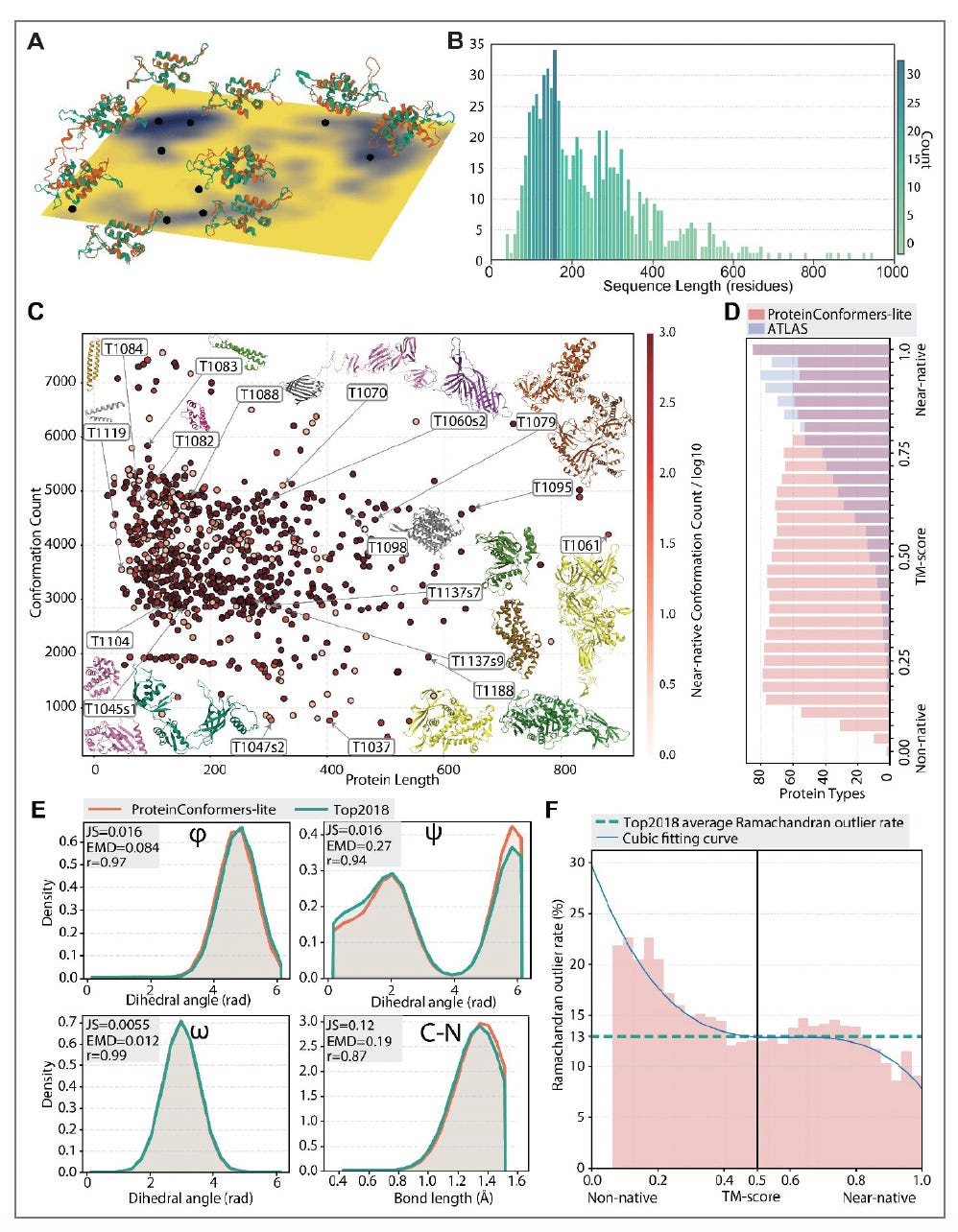
Researchers at the National University of Singapore (Yang Zhang’s group) present ProteinConformers, a database of 2.7 million geometry-optimised conformations across 734 proteins, paired with energy evaluations and a benchmarking framework for multi-conformation generators.
🧬 The dataset uses a multi-seed decoy sampling strategy: for each protein, hundreds of diverse starting conformations (drawn from CASP5-15 prediction submissions) are each run through full-atom molecular dynamics simulation using GROMACS 2023 with the OPLS-AA force field. Each conformation receives five energetic evaluations (RW, RWplus, EvoEF2, Rosetta, FoldX) and pairwise similarity annotations (TM-score and RMSD). The curated benchmark subset, ProteinConformers-lite, contains 381,546 MD-refined conformers across 87 CASP14/15 proteins with 1.9 million energetic annotations.
⚡ ProteinConformers spans protein lengths from 33 to 949 residues, with conformations distributed continuously from non-native to near-native states (TM-scores covering the full 0 to 1 range). Local geometric quality matches the Top2018 reference set: dihedral angle distributions show Pearson correlations of 0.97 to 0.99, and near-native conformations fall below the Top2018 average Ramachandran outlier rate of 13%. In benchmarking five generative models, BioEmu achieves the highest coverage under strict energy thresholds (5 kJ/mol), while AlphaFlow-MD scores comparably on the CGMSmah geometric plausibility metric. Total compute cost: approximately 40 million CPU hours.
🔬 Applications and Insights
1️⃣ Benchmarking Conformation Generators
ProteinConformers-lite is the first standardised evaluation framework for models like AlphaFlow, ESMFlow, and BioEmu. It measures both diversity (how much of the energy landscape a model covers) and plausibility (geometric realism) in one benchmark.
2️⃣ Allosteric Mechanism Studies
The continuous energy surfaces from non-native to native states let researchers study allosteric transitions and functionally relevant intermediates that single-seed MD simulations would miss.
3️⃣ Drug Discovery Applications
Energy-annotated conformational ensembles support ensemble docking workflows, where sampling multiple receptor states improves virtual screening hit rates compared to docking against a single crystal structure.
4️⃣ Evaluating Energetic Realism
With five energy functions evaluated per conformation, the dataset allows systematic comparison of how well different generators produce physically plausible low-energy structures versus merely diverse ones.
💡 Why This Is Cool
If you want to compare AlphaFlow against BioEmu against ESMFlow, you currently have no standard reference to test against. ProteinConformers fills that gap. Every conformation has known energy and measured structural similarity to the native state, and the web platform lets you explore without running simulations.
📄 Read the paper
💻 Explore the database
🗓️ Events & Competitions
The best competitions, hackathons, and community challenges in AI x life sciences, curated weekly. Know something worth featuring? Reply and let us know.
More upcoming events:
London Protein Design Day | June 23, Imperial College London
The first edition of a one-day symposium bringing together London’s protein design community and beyond. Programme spans AI-driven design, molecular dynamics, and bioinformatics, with applications across enzymes, antibodies, and materials. Organised by Pietro Sormanni, Rebecca Birolo, and Jakub Lála. Abstract deadline for poster/oral presentations is this Saturday (May 17). In person only.
BioHackathon Europe 2026 | November 9-13, Barcelona
ELIXIR’s annual international bioinformatics hackathon, running since 2018. 160+ participants, five days of collaborative coding on open bioinformatics infrastructure and tools. The call for project proposals opens March 16 and closes April 15 - so if you want to lead a project, that’s your window.
Thanks for reading!
💬 Get involved
We’re always looking to grow our community. If you’d like to get involved, contribute ideas or share something you’re building, fill out this form or reach out to me directly.
Connect With Us
Have questions or suggestions? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website