
⬡ SureChEMBL 2.0: Rebuilding Patent Chemistry for the AI Era
Deep Dive | Edition 14

Welcome back to the deep dive, where we break down the AI tools and data reshaping how new drugs are discovered. In each edition, we speak directly with the teams behind these tools to explain what they solve, how they work and where they are going next.
What’s your biggest time sink in early drug discovery process?
This week we’re diving into SureChEMBL 2.0, a major redesign of one of the most important open datasets in drug discovery: the global map of chemical structures extracted from patents.
SureChEMBL today contains more than 31 million patent-derived compounds collected from international patent offices. As the scale of patent chemistry increased, the original system began to reach its limits. To understand how the rebuild took shape, we spoke with Nicolas Bosc, Senior Scientific Developer at EMBL-EBI.
🔴 The Problem
There is no global place to access patents. They are made available daily from the different patent authorities (USPTO, EPO, WIPO, JPO etc.) usually as XML, HTML or as text/image PDFs. Heterogeneity also translates in the languages used by the different authorities. Moreover, these resources lack a systematic and continuous annotation mechanism, and full-text searching capabilities. SureChEMBL was introduced to solve these issues by providing a central place for accessing patent documents in a standard human-readable format, and chemically annotated. However, the original SureChEMBL pipeline was built on ageing components and a monolithic architecture. Users increasingly wanted biological context, but the system had been engineered to extract only chemical structures.
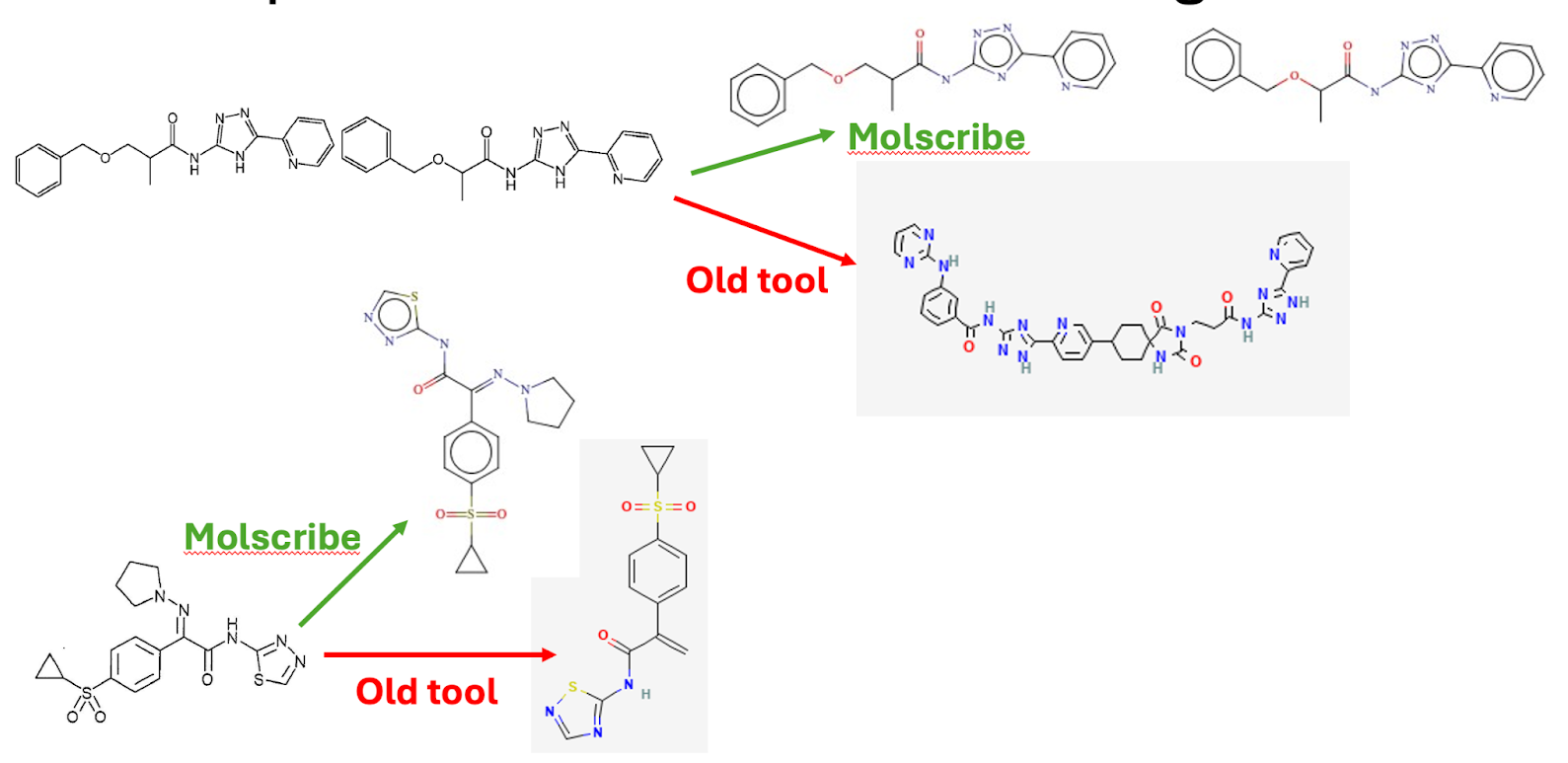
A critical challenge was the nature of patent chemistry itself. A recent analysis in Scientific Data confirmed that almost half of all structures in SureChEMBL originate from images rather than text. This meant that image-to-structure performance was one of the most important factors, as exemplified in another analysis. When structures were drawn close together or at inconsistent quality, older optical recognition tools sometimes merged them into a single incorrect molecule, weakening downstream analysis.
Also USPTO molfile attachments often contain inconsistencies, with some structures far from reality.
In a fully automated pipeline, robust quality control becomes crucial, but in an ageing system this had become a challenge.The combination of noisy inputs, fragile tooling and growing scientific expectations made a full rebuild inevitable.
💡 The Idea
SureChEMBL 2.0 is the result of a multi-year modernisation effort focused on scalability, transparency and scientific strength.
The new system runs on Kubernetes for orchestration and Kafka for handling real-time data streaming, replacing components that were difficult to monitor and prone to failure. Chemical registration and normalisation have been rebuilt using RDKit, providing explicit, reproducible rules in place of legacy logic. This is especially important for organisations using SureChEMBL in medicinal chemistry and machine learning.
The refactor also allowed the team to clarify long-standing assumptions about chemical acceptance and rejection criteria. With millions of structures flowing through the system, clear rules are critical for data quality.
One of the most significant upgrades is the addition of biomedical entity annotation. Historically, SureChEMBL focused purely on chemistry, despite patents containing rich biological information. Early tests with NLP and large language models struggled to produce reliable mappings to ontologies such as UniProt or the Gene Ontology. The breakthrough came from integrating LeadMine, a dictionary-based annotator widely used in chemical text mining, which enabled rapid annotation of the entire database.
📊 Inside the New Pipeline
The updated workflow begins with text annotation for both chemical and biological entities. Work on image is still on-going but using a new generation of optical structure recognition models shows promising results . Molscribe, developed at MIT, showed substantial gains during internal validation, particularly for crowded or low-quality chemical diagrams.
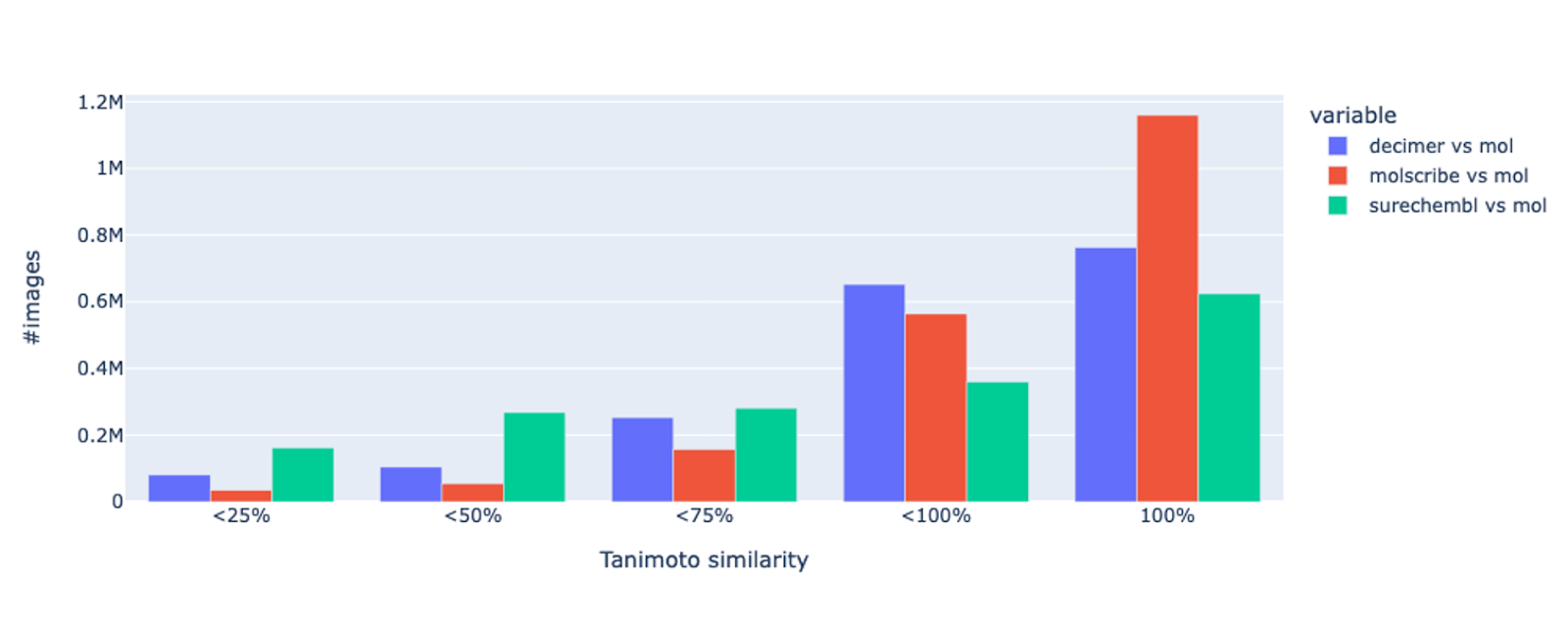
To confirm their first observations, the SureChEMBL team extracted structures from USPTO 2023 patents and compared them with the corresponding molfiles using Tanimoto similarity.
✨ What Is New for Users
The improvements in SureChEMBL 2.0 are immediately visible. The user interface now loads patents within seconds rather than minutes. Bulk downloads, one of the most requested features, have returned and are now available every two weeks. Many organisations ingest the data directly into internal systems and rely heavily on these full releases.
Biomedical annotations introduce a layer of biological insight that previously required manual curation. Although compounds are not yet linked directly to targets or diseases, the annotations allow new workflows, filtering patented structures by disease mentions or exploring chemical space associated with specific genes.
Chemical data quality has also improved. RDKit normalisation, clearer registration logic and more accurate image extraction support QSAR modelling, scaffold exploration and generative chemistry tools, all of which rely heavily on consistent and well-defined structures.
🧬 Why It Is Different
SureChEMBL 2.0 marks a shift from a chemistry-only database to a richer, biologically informed resource. The data are cleaner, the extraction pipeline is more robust, accurate and the architecture is capable of supporting new scientific layers that were previously out of reach. Patent chemistry remains one of the most diverse and challenging sources of novel compounds, and the new pipeline is better equipped to capture that diversity faithfully.
🔮 The Future
The next major challenge is bioactivity extraction. Potency values and assay data often appear in patents long before publication in the scientific literature, but extracting them reliably is not straightforward.
As Nicolas noted, activity data are frequently presented in ways that make it difficult to link a specific compound to the protein tested and the resulting measurement. Targets, assays and numerical values are often dispersed or described ambiguously, reflecting the legal structure of patent writing rather than analytical clarity.
The team plans to explore this area further. If successful, SureChEMBL could unify structures, targets, diseases and activities across the global patent landscape, creating one of the most valuable open datasets for AI-driven drug discovery.
📄 Read the original paperhttps://pmc.ncbi.nlm.nih.gov/articles/PMC4702887/!
📄 Read the SureChEMBL 2.0 announcement.
⚙️ Check out the model.
👨🔬 Get in touch with Nicolas.
📢 Nicolas is presenting SureChEMBL2.0 at the Cambridge Cheminformatics Meeting next week! Check it out.
Thanks for reading Kiin Bio Weekly!
💬 Get involved
We’re always looking to grow our community. If you’d like to get involved, contribute ideas or share something you’re building, fill out this form or reach out to me directly.
Subscribe now to stay at the forefront of AI in Life Science and keep up with this upcoming season of deep dives.
Connect With Us
Have questions on this or suggestions for our next deep dive? We’d love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website
Thank you for the summary. SureChEMBL is a great resource, and I had missed the fact that it has been updated