
Toronto's C3P, Tsinghua's BioMatrix, and Surrey's JEDEL
Kiin Bio's Weekly Insights

Welcome back to your weekly dose of AI news for Life Science! This weeks fix:
C3P uses proteins as a training signal to learn promoter representations, which lets you find co-regulated genes across bacterial genomes without any experimental data. Simple idea, works well.
BioMatrix is another biological foundation model, but this one actually covers the full matrix: molecule sequences, molecule structures, protein sequences, protein structures, and natural language, all in one model. State-of-the-art or competitive on 77 of 80 benchmarks.
JEDEL automates DNA-encoded library design from a pharmacophore, and everything it generates is synthesisable from purchasable building blocks. That constraint is what makes it useful rather than academic.
Kiin Pioneer Programme
We built a platform that helps researchers speed up their entire science, from literature review and biomarker discovery to bioinformatics and computational chemistry. If your workflow involves pulling findings from five different places before you can actually act on any of them, this is for that.
The Pioneer Programme gives academic labs and non-profits one year of free access, plus support from our science team. No cost, no data transfer, all IP stays with your institution. Applications close August, cohort starts September.

C3P: Contrastive promoter-protein pretraining for bacterial gene regulation
🧪 Where This Fits
Genome language models (like DNABERT, Nucleotide Transformer, Evo) have gotten good at learning sequence representations, but they still struggle with regulatory DNA. Promoters are short, non-coding, and their function depends on context that pure sequence models have trouble capturing. The standard approach is to pretrain on raw DNA and hope the model picks up regulatory grammar along the way. It mostly does not.
C3P takes a different angle entirely. Rather than trying to learn promoter function from DNA sequence alone, it uses the protein that a promoter regulates as a supervisory signal. The logic: promoters that regulate similar functions should have similar representations, and protein language models already capture functional similarity well. So you can transfer that knowledge to the promoter side through contrastive learning. It is a simple idea, and arguably obvious in hindsight, but nobody has done it at this scale before.
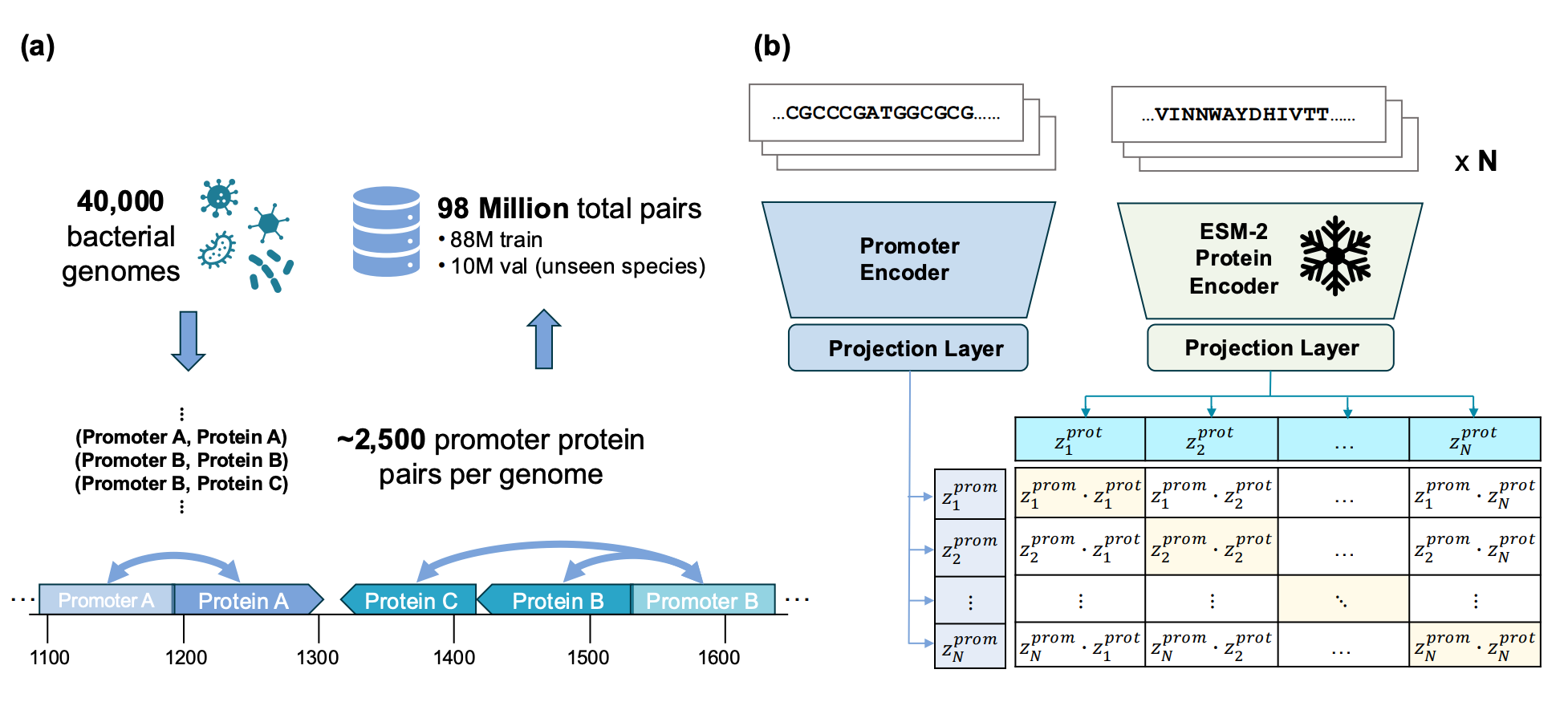
🔍 What It Is
Genome language models learn poor representations of regulatory DNA, limiting their use for predicting gene regulation. Dufault, Xu, and Moses from the University of Toronto present C3P, a contrastive learning framework that pairs bacterial promoters with their downstream proteins to learn promoter representations.
Trained on 88 million promoter-protein pairs using a CLIP-style objective. The promoter encoder learns to align with embeddings from a frozen protein language model, so promoters regulating functionally similar proteins end up with similar representations.
Multi-fold improvement over leading genome language models on regulatory annotation prediction. Enables zero-shot co-regulated gene retrieval: given a promoter, find other promoters driving similar functions, across genomes, with no experimental data required.
💡 Why This Is Cool
The clever bit here is recognising that we already have a strong signal for regulatory function, it just lives in protein space rather than DNA space. Protein language models have encoded functional relationships that took decades of biochemistry to establish. C3P bridges that knowledge back to the regulatory side. This matters for microbiology because most bacterial genomes have no experimental regulatory data at all. If this approach generalises beyond the training distribution (which the zero-shot retrieval results suggest it might), it opens up regulatory annotation for millions of uncharacterised organisms. The limitation is that it only works where you have a clear promoter-protein pair, which excludes non-coding RNAs and complex eukaryotic regulation.
📃 Read the paper.
💻 Try the code.
BioMatrix: A comprehensive biological foundation model spanning sequences, structures, and language
🧪 Where This Fits
The biological foundation model space has been fragmented. You have protein language models (ESM, ProtTrans), molecule models (MolBERT, ChemBERTa), structure predictors (AlphaFold, ESMFold), and various attempts to combine two of these modalities. What nobody has done convincingly is put all five modalities (molecule sequence, molecule structure, protein sequence, protein structure, and natural language) into one architecture that can both read and generate all of them. Previous multi-modal attempts (BioMedGPT, Galactica) either used modality-specific encoders bolted onto a language model, or covered text plus one other modality.
BioMatrix from Tsinghua and collaborators argues you do not need specialised encoders at all. You tokenise everything into a shared vocabulary, train with standard next-token prediction, and let the model sort out the relationships. The scale helps: 304 billion tokens across all modalities, built on Qwen3 at 1.7B and 4B parameters.
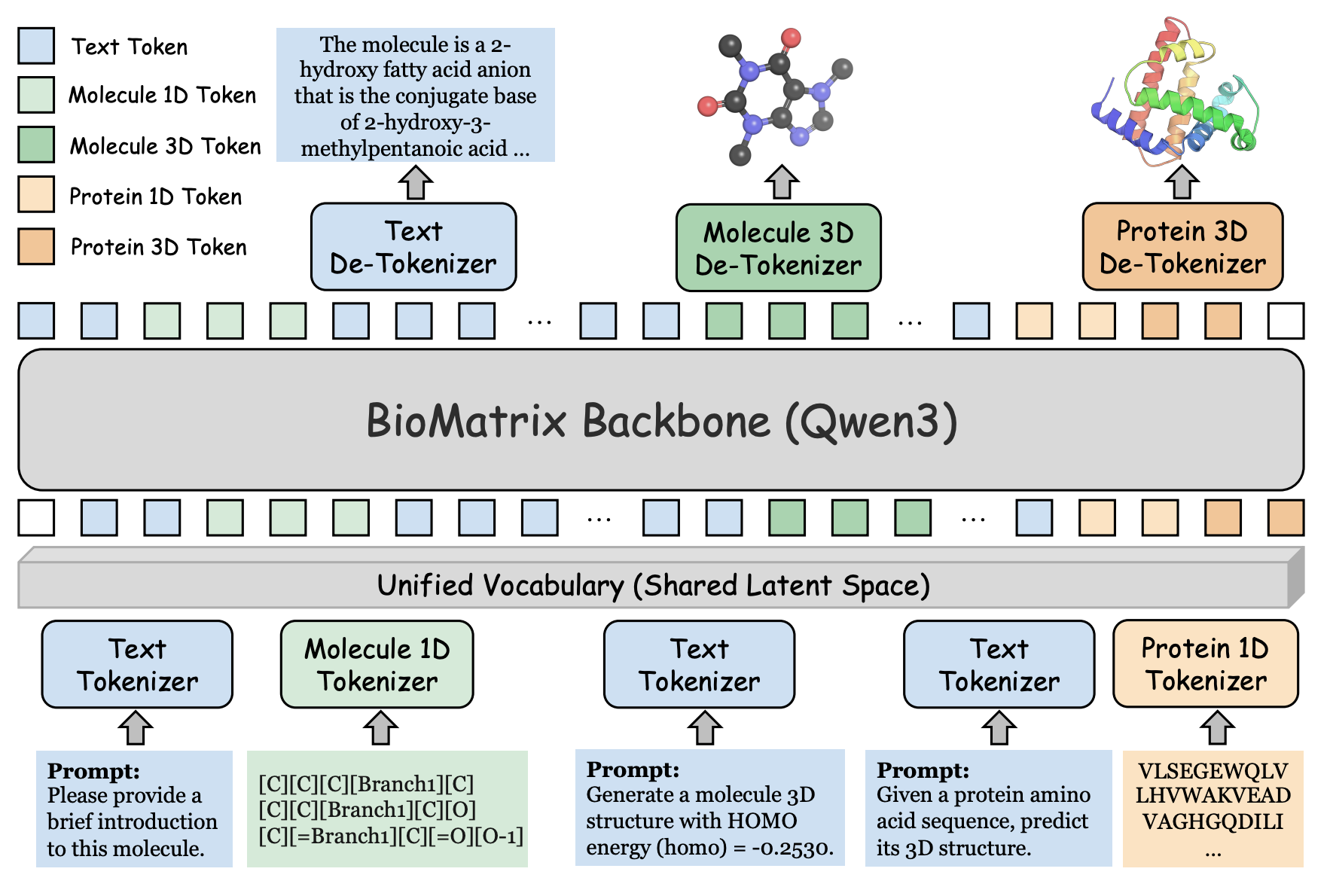
🔍 What It Is
Existing biological AI models are specialised to one or two data types and cannot transfer knowledge between molecules and proteins natively. Pei et al. from Tsinghua present BioMatrix, a multimodal foundation model that unifies molecule sequences, molecule structures, protein sequences, protein structures, and natural language in a single architecture.
All modalities are mapped to a shared token space using learned structure tokenisers (VQ-VAE for molecular and protein 3D structures). The model is then trained with a standard next-token prediction objective, with no external encoders or modality-specific output heads. Built on Qwen3, trained on 304.4 billion tokens.
State-of-the-art or competitive on 77 out of 80 downstream tasks across property prediction, molecule generation, protein folding, captioning, and binding affinity prediction. Outperforms modality-specific specialist models on several benchmarks.
💡 Why This Is Cool
The interesting question is not “does a bigger model do well on benchmarks” (it does, unsurprisingly) but whether unifying modalities in one model creates emergent cross-modal capabilities that specialist models cannot replicate. The paper does not fully answer this yet, and most of the 80 tasks are single-modality evaluations that a specialist could handle. The real test will be tasks that require reasoning across modalities simultaneously: “given this protein structure and this molecule, predict binding and explain why.” If BioMatrix can do that better than a pipeline of specialists, the unified approach is vindicated. If it just matches specialists on their own turf, the value proposition is convenience rather than capability. The Apache 2.0 license and public weights make it easy to test either way.
📃 Read the paper.
💻 Try the code.
JEDEL: Zero-shot DNA-encoded library design for early-stage drug discovery
🧪 Where This Fits
DNA-encoded libraries (DELs) are combinatorial chemistry at industrial scale: you attach DNA barcodes to building blocks, combine them through validated reactions, and screen the resulting millions of compounds against a target. The design problem is choosing which building blocks and reactions to include. Traditional DEL design relies on chemical diversity heuristics or brute-force enumeration, neither of which accounts for whether the resulting library will actually produce binders for your specific target.
Generative drug design models (like those from Recursion, Insilico, etc.) can propose target-specific molecules, but they produce virtual structures that then require separate synthesis planning, which often fails. JEDEL closes this gap by designing libraries that are target-aware and synthesisable by construction: every output is built from purchasable building blocks through validated reactions.
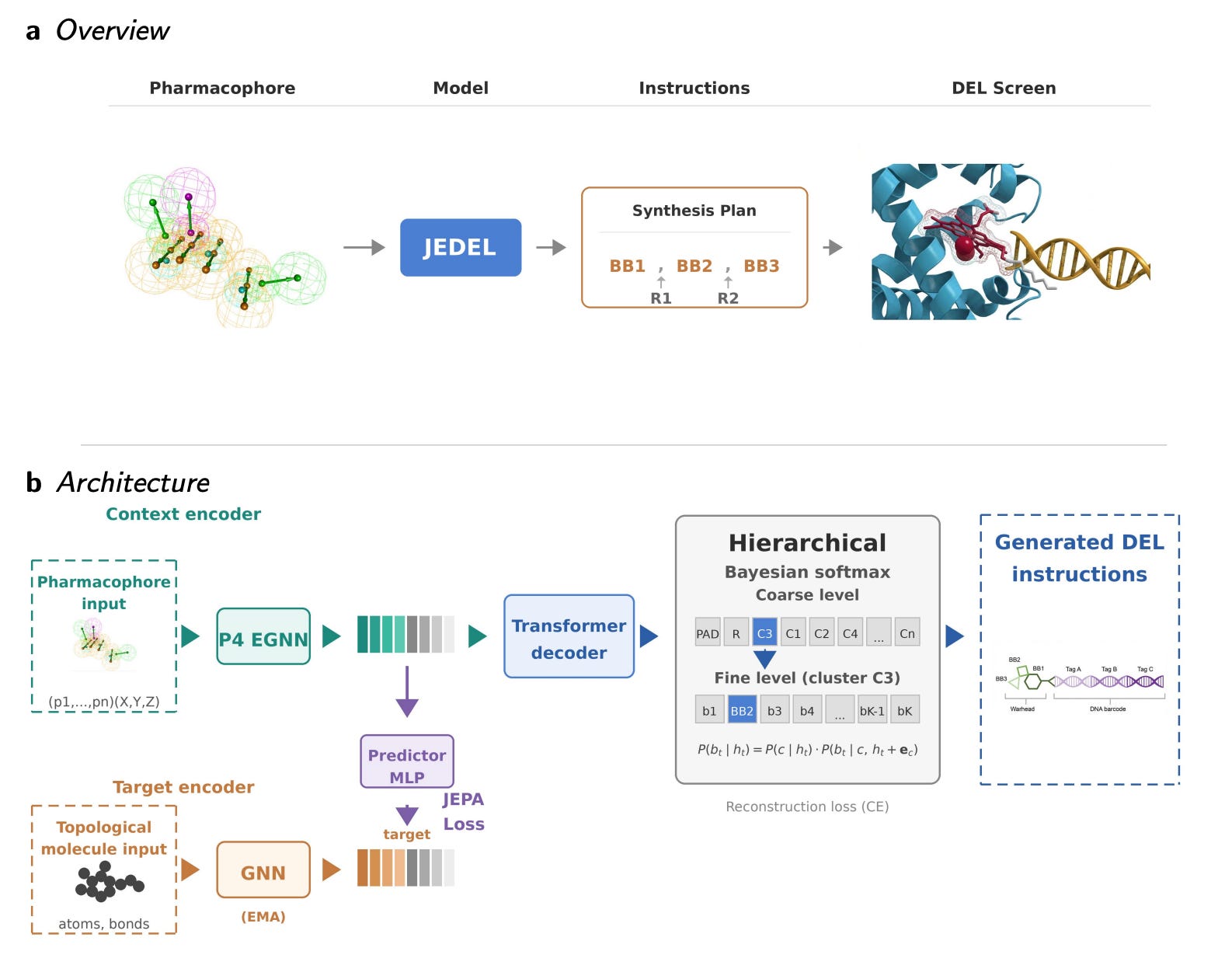
🔍 What It Is
Current DEL design ignores target information, and generative models propose molecules that are often unsynthesisable. Jocys et al. from the University of Surrey present JEDEL, a framework that converts 3D pharmacophore information from known active ligands into synthesis instructions for target-focused DNA-encoded libraries.
Takes pharmacophore representations as input, searches a space of purchasable building blocks and validated combinatorial reactions, and assembles libraries that are synthesisable by definition. Requires no target-specific retraining, operating in a zero-shot manner across different protein targets.
Tested across 18 protein targets, JEDEL outperformed random and diversity-based baselines on predicted binding affinity, pharmacophore recovery, and sample efficiency. The constraint to purchasable reagents means every designed library can be made in the lab immediately.
💡 Why This Is Cool
DELs are already a workhorse in pharma (Novartis, GSK, and X-Chem all run them at scale), so improvements here have a clear commercial path. What JEDEL does is shift library design from “maximise chemical diversity and hope something binds” to “build a library biased toward your target’s pharmacophore.” The zero-shot aspect means you do not need to retrain for each new campaign. The synthesisability constraint is the real differentiator from generative models: the gap between “here is a molecule that might bind” and “here is a library you can make on Monday” is where most computational drug design papers lose their translational value. The limitation is that pharmacophore inputs require existing active compounds, so this is a hit expansion tool rather than a de novo discovery method. For teams already running DEL campaigns, this looks immediately applicable.
📃 Read the paper.
No public code repository available at time of writing.
🗓️ Events & Competitions
The best competitions, hackathons, and community challenges in AI x life sciences, curated weekly. Know something worth featuring? Reply and let us know.
More upcoming events:
BioHackathon Europe 2026 | November 9-13, Barcelona
ELIXIR’s annual international bioinformatics hackathon, running since 2018. 160+ participants, five days of collaborative coding on open bioinformatics infrastructure and tools. The call for project proposals has now closed.
Thanks for reading!
💬 Get involved
We’re always looking to grow our community. If you’d like to get involved, contribute ideas or share something you’re building, fill out this form or reach out to me directly.
Connect With Us
Have questions or suggestions? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website