
TU Eindhoven's STRIPES, Harvard/UIUC's TrialBench, and FSU Jena's ChemAudit
Kiin Bio's Weekly Insights

Welcome back to your weekly dose of AI news for Life Science!
What’s your biggest time sink in the drug discovery process?
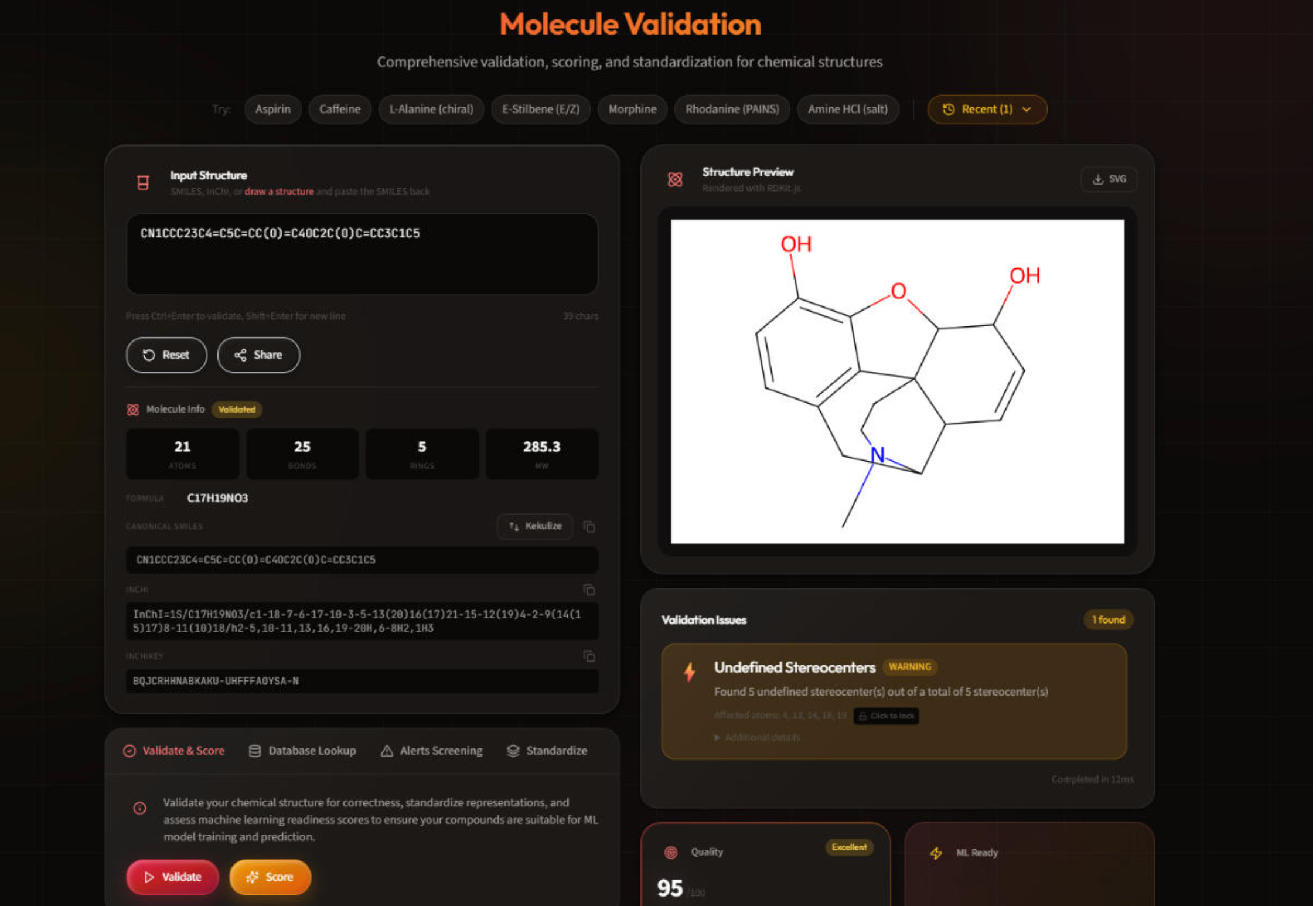
ChemAudit: Open-source chemical structure validation for chemistry ML pipelines
🔬 Bad chemistry data doesn’t announce itself. Incorrect stereochemistry, undefined stereocenters, PAINS-flagged compounds, inconsistent tautomers. These issues don’t throw errors. They just quietly degrade your models, and you find out downstream when it’s already too late.
ChemAudit is a free, open-source web platform consolidating structure validation, standardisation, structural alert screening, and ML-readiness scoring in one interface. No command line required.
🧬 Built on RDKit, MolVS, and the ChEMBL structure pipeline, it runs 15+ validation checks, screens 480+ PAINS patterns and 700+ pharmaceutical filters, and scores ML-readiness from 0-100 across 451 molecular descriptors and 7 fingerprint types. Batch processes up to 1M molecules. MIT-licensed and self-hosted.
⚗️ Applications & Insights
1️⃣ End-to-End Structure Validation
15+ checks across parsability, valence, stereochemistry, and representation consistency catch errors before they propagate into training data or downstream analyses.
2️⃣ Structural Alert Screening at Scale
480+ PAINS patterns and 700+ pharmaceutical filter rules flag problematic compounds, traceable to BMS, Glaxo, Dundee, and ChEMBL, across drug discovery and natural products workflows.
3️⃣ Quantitative ML-Readiness Scoring
Scoring against 451 descriptors and 7 fingerprint types gives an objective measure of dataset quality. Not just structural validity, but actual usability in ML pipelines.
4️⃣ Drug-Likeness and ADMET in One Place
Lipinski, QED, Veber, Ghose, and Muegge alongside ADMET predictions for synthetic accessibility, solubility, and CNS penetration. No switching between tools.
💡 Why This Is Cool
Anyone who has cleaned a chemical dataset knows the fragmented toolchain this replaces. ChemAudit consolidates the entire workflow into one accessible interface backed by the most trusted open-source chemistry libraries. Built for scale and free to use.
📖 Try the tool
💻 Try the code
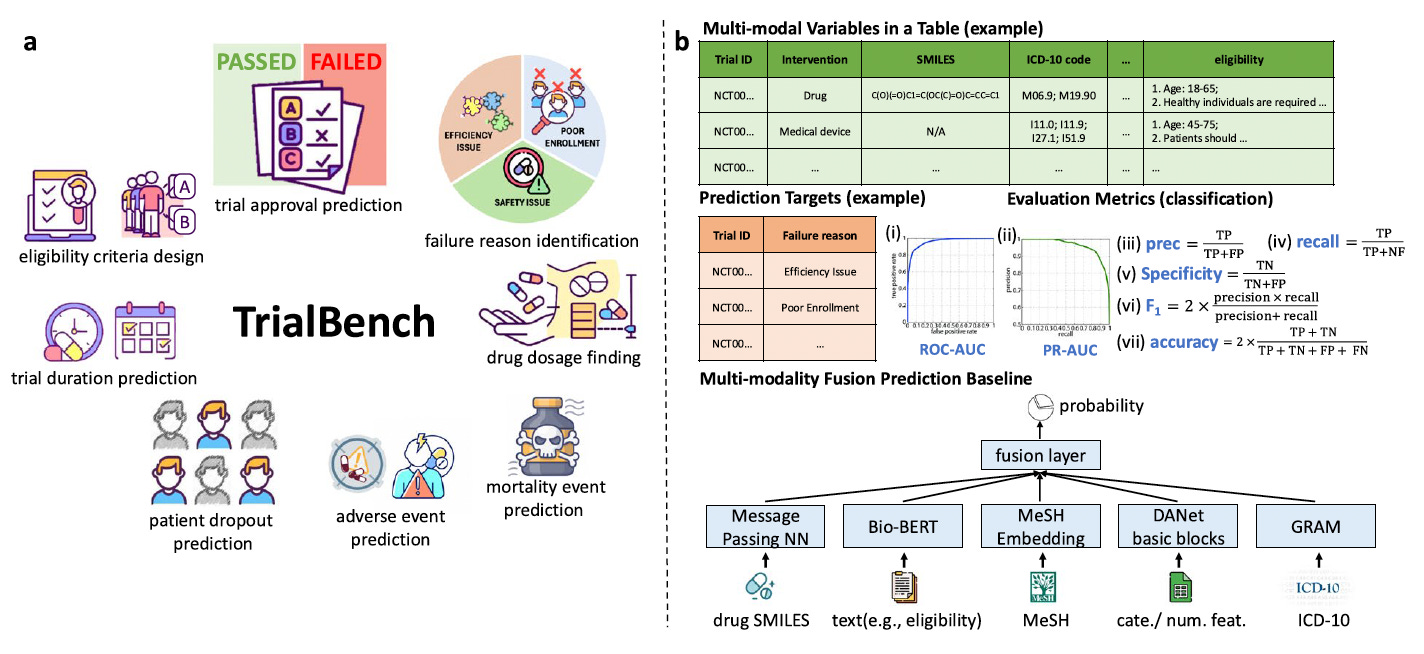
TrialBench: Multi-modal AI-ready datasets for clinical trial outcome prediction
🔬 Over 90% of clinical trials fail. The data that could predict those failures (dropout rates, adverse event patterns, dosing miscalculations, duration overruns) already exists. It’s just fragmented, inconsistently formatted, and nearly impossible for ML researchers to use.
TrialBench changes that. Published in Nature Scientific Data, this benchmark delivers 23 curated, multi-modal, AI-ready datasets spanning eight clinical trial prediction tasks, assembled from ClinicalTrials.gov, DrugBank, TrialTrove, and ICD-10.
🧬 The eight tasks span the full trial lifecycle: duration, dropout, adverse events, mortality, trial approval, failure reasons, eligibility criteria design, and drug dose finding. Each dataset pairs structured metadata, molecular drug representations, and free-text clinical descriptions, the same mix a human reviewer works with.
⚡ Open Python and R packages handle loading and benchmarking, with baseline models and standardised splits for every task, enabling direct comparison without rebuilding evaluation pipelines.
📊 Applications & Insights
1️⃣ Standardised Benchmarking Across the Trial Lifecycle
Eight tasks covering every major failure point, from protocol design to post-trial outcomes, in one resource. No bespoke dataset assembly required.
2️⃣ Multi-Modal Data Integration
Structured metadata, molecular representations, and free-text clinical descriptions come packaged together: the heterogeneous mix real-world prediction actually demands.
3️⃣ Failure Prediction at Multiple Levels
Tasks span early failure modes (dosing, eligibility design) and late-stage outcomes (adverse events, mortality, approval), a framework for understanding where trials break down.
4️⃣ Reproducible Baselines for Fair Comparison
Standardised splits and baseline implementations let new methods be directly compared. No need to rebuild evaluation pipelines from scratch.
💡 Why This Is Cool
Clinical trial prediction has been a graveyard for AI benchmarks: data siloed, endpoints inconsistent, results near-impossible to reproduce. TrialBench addresses all three at once. It won’t solve clinical AI alone, but it gives the field a common language.
📖 Read the paper
💻 Try the code
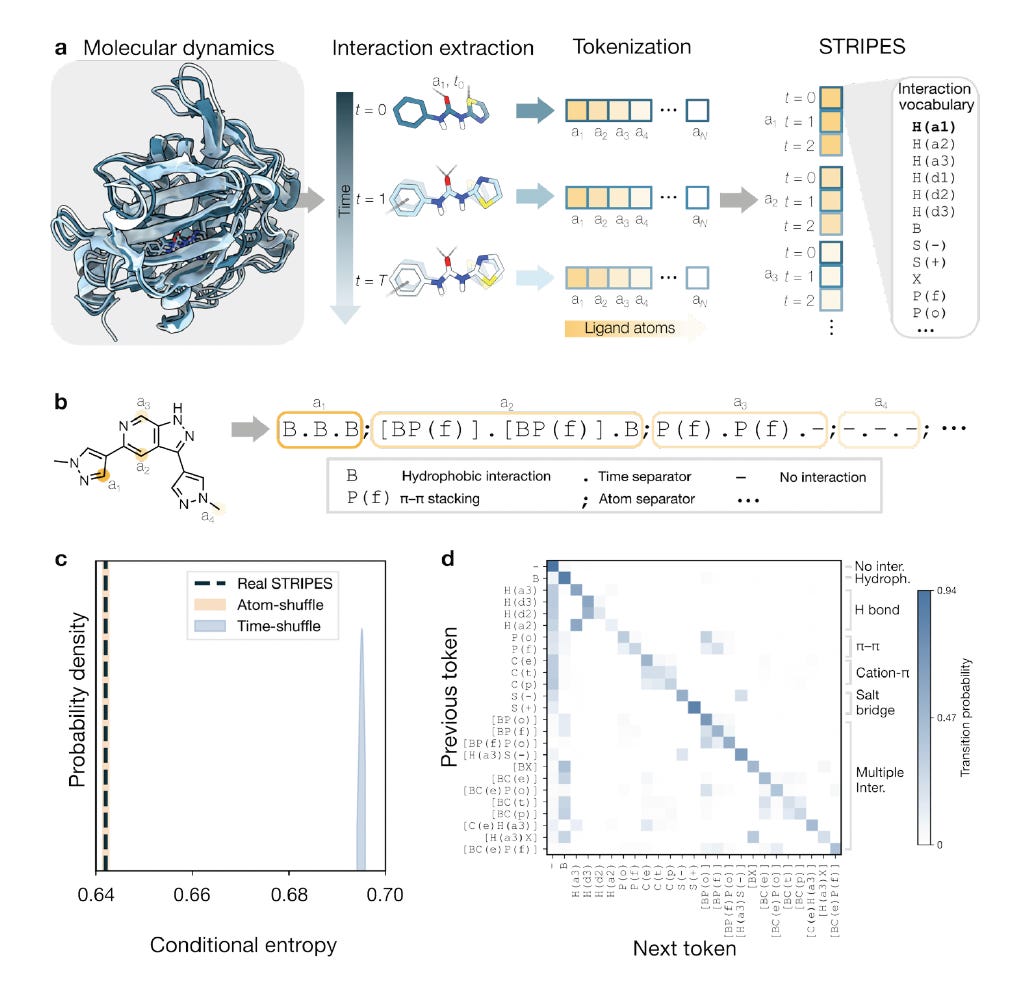
STRIPES: A symbolic language of molecular recognition for structure-informed drug design
🔬 Protein-ligand binding isn’t a static snapshot. It’s a dynamic interplay of interactions that shifts over time. Most drug design tools treat it as one. The information locked inside MD trajectories is largely untapped.
Researchers at Eindhoven University of Technology developed STRIPES, encoding protein-ligand interaction dynamics from MD simulations as symbolic token sequences, treating molecular recognition as a language for retrieval, comparison, and generation.
🧬 STRIPES converts interaction fingerprints: hydrogen bonds, hydrophobic contacts, pi-stacking, electrostatics, into compact symbolic strings over time. These form a physically interpretable record of binding, enabling similarity retrieval and conditioning of de novo generation on target interaction profiles.
⚡ Validated across JAK1, PIM1, PPARd, and the androgen receptor on ~15,000 MD trajectories: STRIPES-guided Transformer design produced confirmed hits at picomolar to nanomolar potency. Conditioning on interaction patterns opens routes conventional methods miss.
🔬 Applications & Insights
1️⃣ Interaction-Conditioned De Novo Generation
STRIPES enables ligand generation conditioned on specific interaction profiles, moving beyond shape and pharmacophore to dynamic binding behaviour captured from real MD trajectories.
2️⃣ Physically Interpretable Representations
Each token maps to a real interaction type. Unlike black-box embeddings, STRIPES remains interpretable, supporting hypothesis-driven design and mechanistic insight into binding.
3️⃣ Trajectory-Level Similarity Search
STRIPES sequences enable fast comparison of binding dynamics across large MD datasets, identifying compounds with similar interaction fingerprints regardless of structural similarity.
4️⃣ Experimental Validation Across Four Targets
Compounds were synthesised and tested on JAK1, PIM1, PPARd, and AR, confirming picomolar to nanomolar activity and going beyond benchmarking to real biological validation.
💡 Why This Is Cool
STRIPES makes MD trajectories a generative resource, not just an analytical one. Encoding binding dynamics as a learnable language adds a physically grounded layer to drug design that static docking can’t offer.
📖 Read the paper
💻 Code
🗓️ Events & Competitions
The best competitions, hackathons, and community challenges in AI x life sciences, curated weekly. Know something worth featuring? Reply and let us know.
Last week we featured two Adaptyv Bio hackathons - check back soon for updates on results.
BioHackathon Edinburgh 2026 | March 20-22, Edinburgh
Three days at the University of Edinburgh bringing together life scientists, programmers, and industry partners to hack on real biological challenges. Tracks cover academic research (gene regulation, drug discovery, imaging), industry innovation, and a non-coder track for experimental design and project management. Applications are closed, but one to watch if you’re at a UK university for next year.
BioHackathon Europe 2026 | November 9-13, Barcelona
ELIXIR’s annual international bioinformatics hackathon, running since 2018. 160+ participants, five days of collaborative coding on open bioinformatics infrastructure and tools. The call for project proposals opens March 16 and closes April 15 - so if you want to lead a project, that’s your window.
Thanks for reading!
💬 Get involved
We’re always looking to grow our community. If you’d like to get involved, contribute ideas or share something you’re building, fill out this form or reach out to me directly.
Connect With Us
Have questions or suggestions? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website