
UCL’s ProFam, TU Berlin’s AS4Km, and Stockholm University’s EvoBind
Kiin Bio's Weekly Insights

Happy New Year and welcome back to your weekly dose of AI news for life science!
What’s your biggest time sink in the drug discovery process?
🧬 ProFam: A Protein Language Model That Learns From Evolution
What if protein language models treated evolution as signal rather than noise?
Most protein language models treat sequences like text, learning patterns without considering the evolutionary families proteins belong to. But proteins do not evolve in isolation. They are shaped by millions of years of selection within families, with shared constraints and functional context.
ProFam, developed by researchers at UCL, NVIDIA and Helmholtz Munich, takes a different approach. It is a family-aware protein language model trained across 40 million protein families, designed to predict fitness, generate new sequences and support structure prediction by explicitly modelling evolutionary context.
Rather than learning from individual sequences alone, ProFam treats sequence generation as a family-level problem. Context matters.
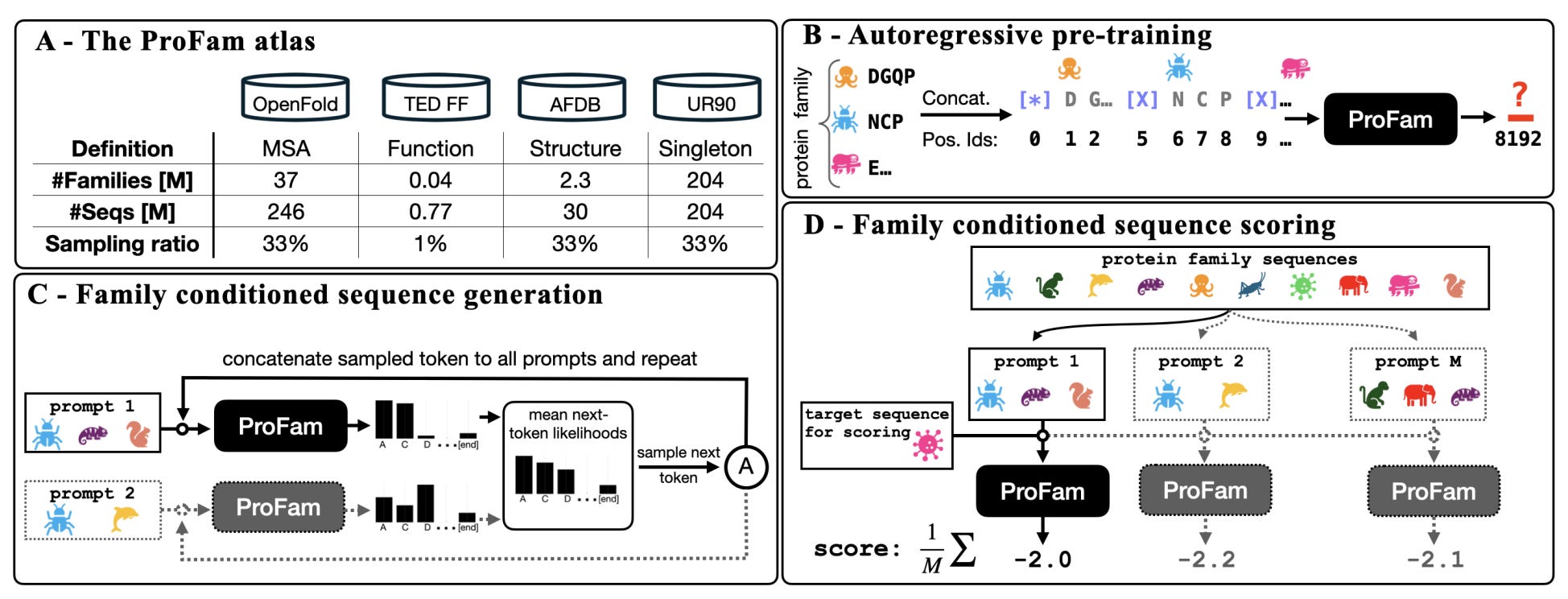
🔬 Applications and Insights
1️⃣ Strong zero-shot fitness prediction
ProFam-1 achieved Spearman correlations of 0.47 for substitutions and 0.53 for insertions and deletions on the ProteinGym benchmark. Performance improves further when predictions are ensembled across prompts.
2️⃣ Faithful sequence generation across enzyme families
Across 460 enzyme families, ProFam outperformed PoET on KL divergence and conservation metrics, even when prompted with a single family member.
3️⃣ Boosting structure prediction with synthetic MSAs
When used to generate synthetic MSAs for ColabFold, ProFam improved lDDT scores from around 0.5 to 0.7 compared with single-sequence inputs. While not matching natural MSAs, the gains are substantial given the lack of alignment data.
4️⃣ Fully open-source release
Model weights, the ProFam Atlas and all training pipelines are openly available, with no API gating or access restrictions.
💡 Why It’s Cool
ProFam leans into the messy reality of evolutionary biology instead of working around it. By learning directly from protein families, it gains range across fitness prediction, sequence design and structure support. It is not just another pLM, but a framework for evolution-aware protein modelling.
📄 Read the preprint
⚙️ Explore the code
🧪 AS4Km: Predicting Enzyme KM by Focusing on the Active Site
What if a simple model could beat state of the art by knowing where the action is?
Predicting KM is deceptively difficult. Experimental data are noisy, measurements vary, and most machine learning models treat enzymes and substrates as separate entities, hoping their interaction emerges later in the network.
Researchers at TU Berlin take a more direct approach. AS4Km explicitly encodes enzyme–substrate interaction interfaces using active site annotations. With this focused representation, even a shallow multilayer perceptron reaches state-of-the-art performance.
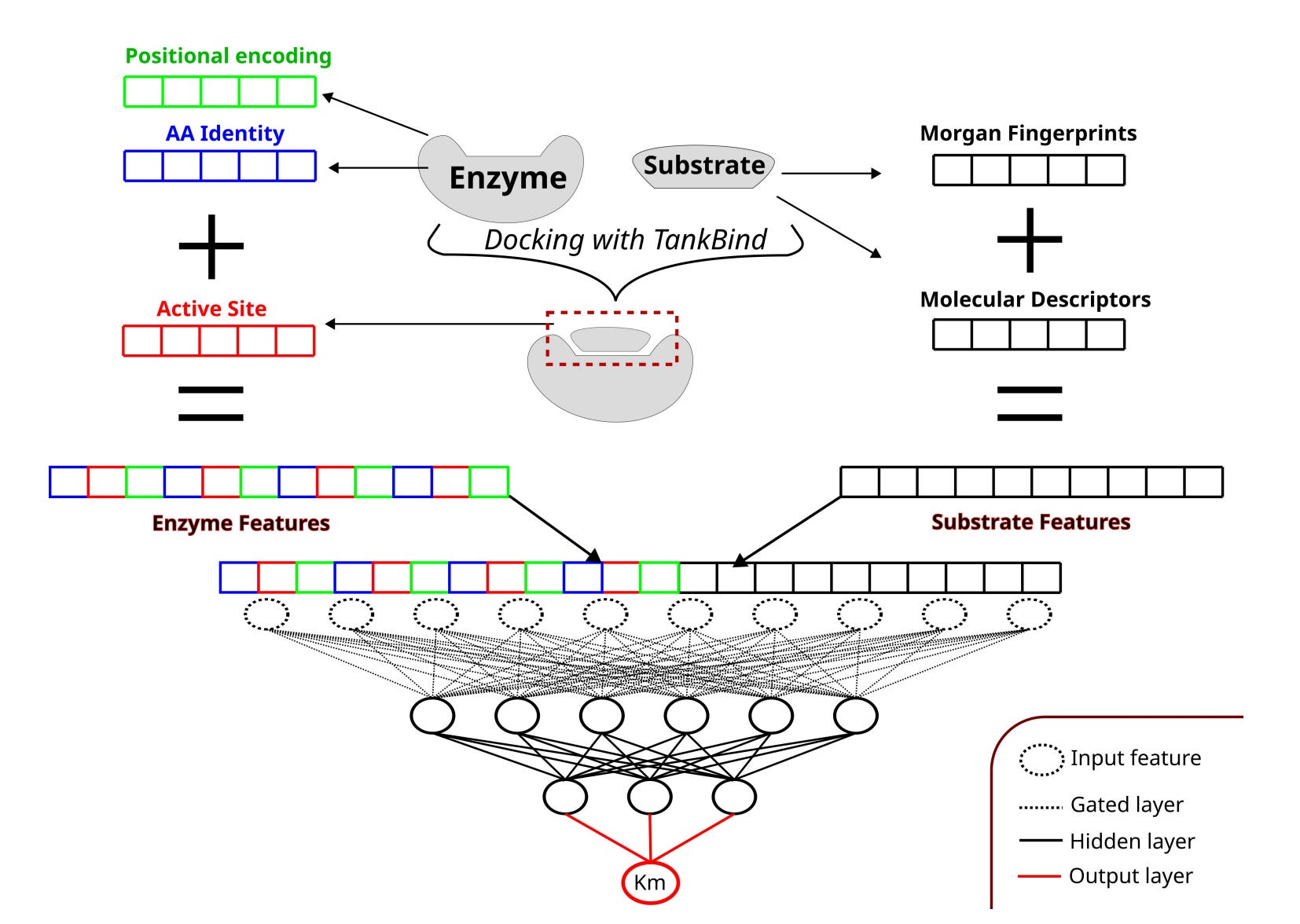
🔬 Applications and Insights
1️⃣ Active site encoding improves generalisation
Using a shallow MLP with P2Rank-predicted binding pockets, AS4Km achieved a Pearson correlation of 0.585 on the HXKm test set, matching or exceeding much deeper models such as GraphKM.
2️⃣ Less sequence, more signal
Ablation studies showed that models using only active site residues outperformed those using the full enzyme sequence. Removing irrelevant context reduced noise and improved predictions.
3️⃣ Uncertainty-aware predictions
By training an ensemble of ten models and preserving KM variance during training, AS4Km captures both epistemic and aleatoric uncertainty, highlighting where predictions are reliable and where they are not.
4️⃣ Open-source and lightweight
The full pipeline is openly available and runs without GPUs. It combines TankBind and P2Rank for annotation with a simple, efficient training loop.
💡 Why It’s Cool
AS4Km shows that progress does not always require larger models. Sometimes, the key is giving a model the right slice of biology. Explicitly encoding the interaction interface turns a simple architecture into a powerful predictor.
📄 Read the preprint
⚙️ Try the code
⚛️ EvoBind: Designing an HIV Inhibitor Without Structural Data
What if you could design a functional inhibitor without knowing the structure or binding site?
Designing inhibitors usually relies on detailed structural information and extensive screening. But researchers at Stockholm University, SciLifeLab and the Karolinska Institute demonstrate that this is not always necessary.
Using EvoBind, a structure-aware AI model, the team designed a single cyclic peptide targeting HIV’s gp41 fusion protein using sequence information alone. No prior binding site, no structural data, and no screening. One design, first attempt, and it worked.
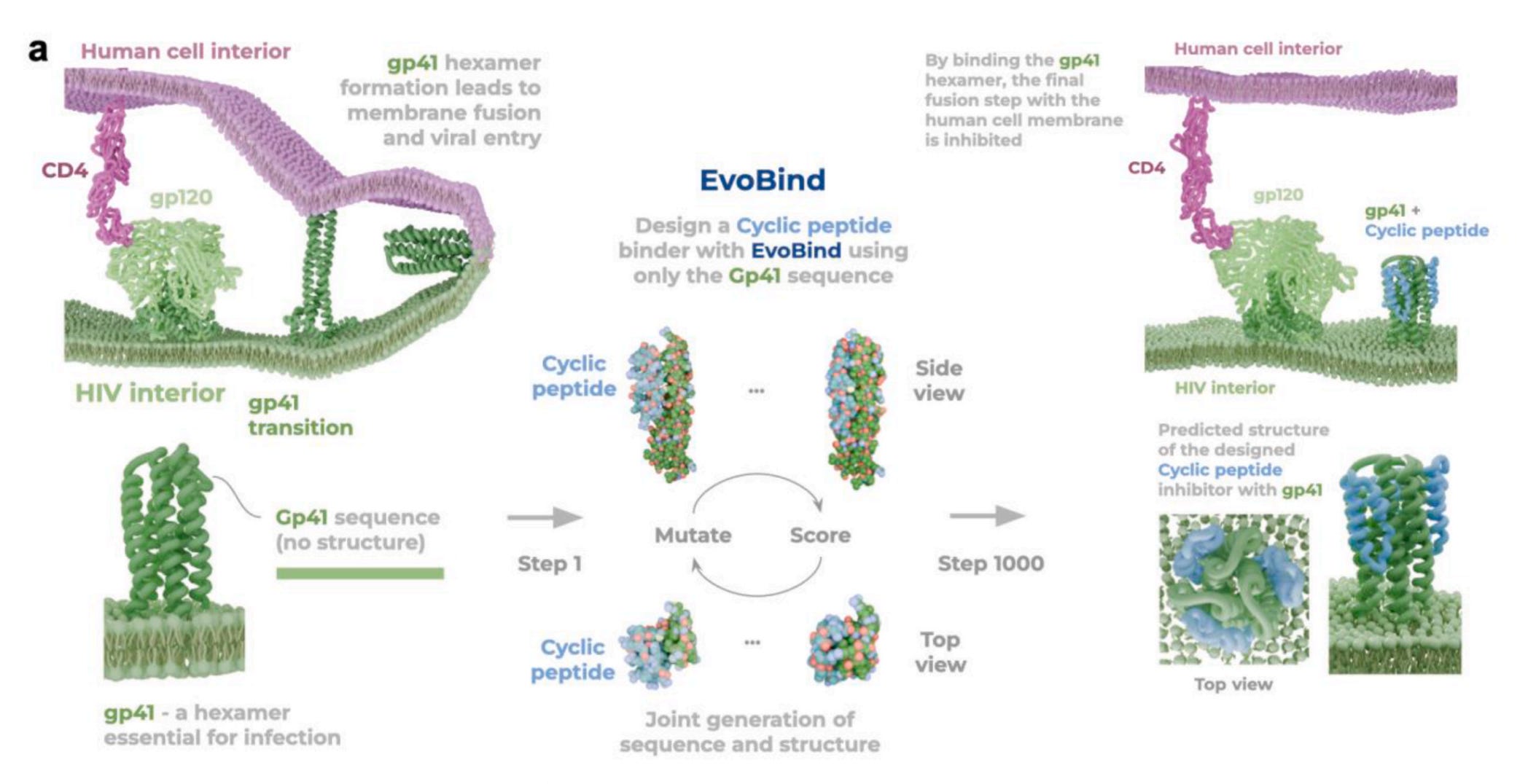
🔬 Applications and Insights
1️⃣ Designed from sequence alone
Using only the gp41 amino acid sequence, EvoBind generated a 39-residue cyclic peptide that binds the transient fusion state of gp41, a conformation exposed for only milliseconds during viral entry.
2️⃣ Potent cellular inhibition
The peptide inhibited replication of HIV-1 strains NL4-3 and AD8 with IC50 values of 9.40 μM and 6.18 μM, with no observed toxicity even at high concentrations.
3️⃣ Biophysical validation
Surface plasmon resonance experiments confirmed binding to the gp41 pseudo-dimer, with a measured Kd of 1.12 μM.
4️⃣ Avoids resistance-prone regions
The peptide targets a conserved interface distinct from known resistance hotspots such as the enfuvirtide binding site, improving prospects for long-term efficacy.
💡 Why It’s Cool
This was not a library screen or a multi-round optimisation. It was a single design generated in 24 hours that targeted an invisible viral conformation and worked. EvoBind shows how AI can enable fast, adaptable responses in settings where time and data are limited.
📄 Read the paper
⚙️ Explore the code
Thanks for reading Kiin Bio Weekly!
💬 Get involved
We’re always looking to grow our community. If you’d like to get involved, contribute ideas or share something you’re building, fill out this form or reach out to me directly.
Connect With Us
Have any questions or suggestions for a post? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website