
University of Copenhagen’s AF-CALVADOS, CASP’s Distance-AF, and Max Planck’s AlphaDIA
Kiin Bio's Weekly Insights

Welcome back to your weekly dose of AI news for Life Science!
What’s your biggest time sink in the drug discovery process?
🧩 AF-CALVADOS: Bringing motion to AlphaFold’s still images.
Simulating proteins that are part folded, part disordered has always been complicated.
AlphaFold predicts structure beautifully, and coarse-grained models like CALVADOS simulate disorder and motion.
Until now, combining the two and doing it across thousands of proteins has been nearly impossible without manual domain mapping and heavy compute.
That’s what AF-CALVADOS, from the University of Copenhagen, changes.
It’s a hybrid simulation framework that automatically integrates AlphaFold’s static predictions with CALVADOS’ dynamic modelling. The result is a scalable, automated way to explore how order and disorder coexist in real proteins.

🔬 Applications and Insights
1️⃣ Proteome-scale simulation
Mapped 12,483 human proteins directly from AlphaFold predictions to CALVADOS simulations with no manual setup.
2️⃣ Confidence-driven motion
Translated pLDDT and PAE scores into positional restraints that define how strongly a region should fold or move.
3️⃣ Experimental validation
Reproduced chain expansion and AlphaFold error matrices, confirming that simulated motion matches physical behaviour.
4️⃣ Open and automated
Generates a public resource for modelling proteins with mixed structure and disorder, from individual domains to the full proteome.
💡 Why It’s Cool
AlphaFold predicts what a protein looks like.
AF-CALVADOS explores how it moves.
It reuses AlphaFold’s “confidence” not as a score but as a force, deciding which parts of a protein stay rigid and which bend or unfold in motion.
That simple inversion turns static predictions into dynamic simulations.
It means we can now watch proteins breathe, fluctuate and reorganise in silico, capturing the real physical complexity of biology at the proteome scale.
📍 The immediate win is automation: no more manual domain setup.
🌍 The deeper win is perspective: understanding the proteome not as a library of static shapes, but as a living landscape of motion.
📄Check out the paper!
⚙️Try out the code.
🧬 Distance-AF: Making AlphaFold move, not just predict
Distance-AF combines human intuition with AI prediction to create dynamic, interactive protein models guided by real experimental data.
AlphaFold2 transformed structure prediction, but it still produces a single static model. Real proteins shift between multiple conformations, and sometimes a few experimental hints can help reveal them.
Distance-AF integrates user-defined distance constraints directly into AlphaFold2. These can come from crosslinking, FRET, cryo-EM density maps, or even hypotheses about residue interactions. By feeding these constraints back into AlphaFold, the system explores conformations that are both physically possible and consistent with experimental data.

🔬 Applications and Insights
1️⃣ Modeling GPCR
GPCRs transition between active and inactive states. AlphaFold2 captures only one, but Distance-AF allows researchers to model functionally relevant conformations, which can improve drug design for these receptors.
2️⃣ Capturing flexible and disordered regions
Long loops, unstructured tails, and linkers often fold unpredictably. Distance-AF preserves the stable core and uses distance restraints to guide flexible regions toward more realistic geometries.
3️⃣ Integrating human hypotheses
Scientists can encode hypothetical residue interactions to test structural ideas in silico before running new experiments.
4️⃣ Fitting models into cryo-EM maps
Cryo-EM derived restraints help refine AlphaFold predictions, producing atomic models that align more accurately with density data for large complexes.
💡 Why It’s Cool
On 25 challenging protein targets, applying just six distance constraints per structure improved AlphaFold2 predictions by an average of 11.75 Å RMSD.
The real innovation lies in how it connects human reasoning with AI.
Distance-AF transforms AlphaFold from a static predictor into a collaborative modeling tool, where researchers can guide the AI to explore alternative conformations and test ideas interactively.
🧠 It is a smarter, more flexible way to model proteins, one that blends human insight with computational precision to better capture how proteins truly behave.
📄Check out the paper!
⚙️Try out the code.
🧬 AlphaDIA: Deep Learning for Feature-Free, Transferable Proteomics
Data-independent acquisition (DIA) transformed proteomics throughput, but data analysis has lagged behind. Traditional pipelines rely on hand-tuned parameters and feature extraction, creating bottlenecks and limiting transfer between instruments.
Researchers at the Max Planck Institute of Biochemistry and the University of Copenhagen built AlphaDIA, a deep-learning framework that reads DIA data directly from the raw signal.
No peak picking, no feature extraction — just direct interpretation of the detector output.
It connects raw ion traces to peptide identities through neural scoring and transfer learning, creating a faster, more accurate, and more adaptive DIA analysis pipeline.
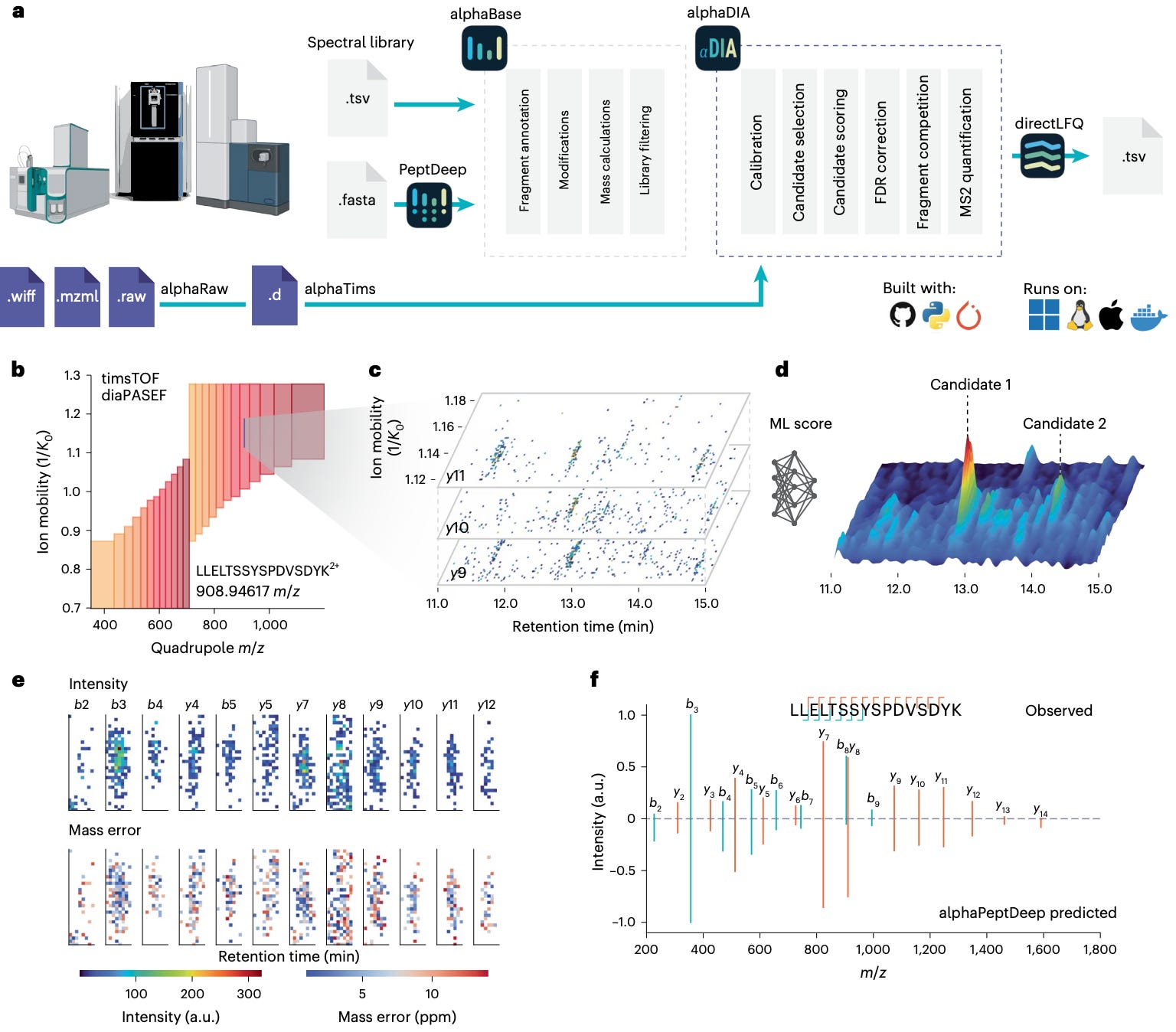
🔬 Applications and Insights
1️⃣ Feature-free peptide identification
AlphaDIA interprets raw DIA signals directly, aggregating data across retention time, ion mobility, and m/z to identify peptides without predefined features.
2️⃣ Cross-platform generalization
Works seamlessly on timsTOF, Orbitrap Astral, and ZenoTOF data, adapting to different acquisition schemes through a unified learning model.
3️⃣ DIA transfer learning
Fine-tunes predicted spectral libraries from AlphaPeptDeep to each instrument and modification type, dramatically improving spectral correlation and retention-time prediction.
4️⃣ Discovery at scale
Identified 73,000 precursors and 6,800 protein groups in 21-minute HeLa runs and achieved a 48% gain in modified peptide detection using transfer learning.
💡 Why It’s Cool
AlphaDIA brings intelligence directly into the proteomics pipeline.
Instead of predefining what to look for, it learns patterns in the raw signal itself, then transfers that knowledge across datasets, instruments, and conditions.
It’s a foundation model for mass spectrometry: open, fast, and adaptive.
A system that learns from the data, not just processes it.
📄Check out the paper!
⚙️Try out the code.
Thanks for reading!
Did you find this newsletter insightful? Share it with a colleague!
Subscribe Now to stay at the forefront of AI in Life Science.
Connect With Us
Have questions or suggestions? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website