
UT Dallas’ ChemRefine, Merck + NVIDIA’s KERMT, and MIT + UW’s Protein Hunter
Kiin Bio's Weekly Insights

Welcome back to your weekly dose of AI news for Life Science!
What’s your biggest time sink in the drug discovery process?
Protein Hunter: Turning Diffusion Hallucination Into a Protein Design Strategy
Most protein design tools either optimise endlessly or rely on heavy fine-tuning. They can generate new folds, but usually only after a lot of compute, filtering, and (really) luck.
Protein Hunter, a new framework from MIT and the University of Washington, takes a different approach. It harnesses the natural hallucination ability of diffusion-based structure predictors like AlphaFold3 to create new proteins from scratch. So, no retraining, no gradients, no prior structure. Sounds pretty good.
Starting from an entirely blank sequence, just a string of “X” tokens, the model hallucinates a folded backbone, which is then refined through iterative cycles of ProteinMPNN sequence design and structure prediction. Each cycle strengthens foldability, stability, and secondary structure until the protein converges to a realistic design. Essentially, its structure and sequence learning to fit each other in real time.
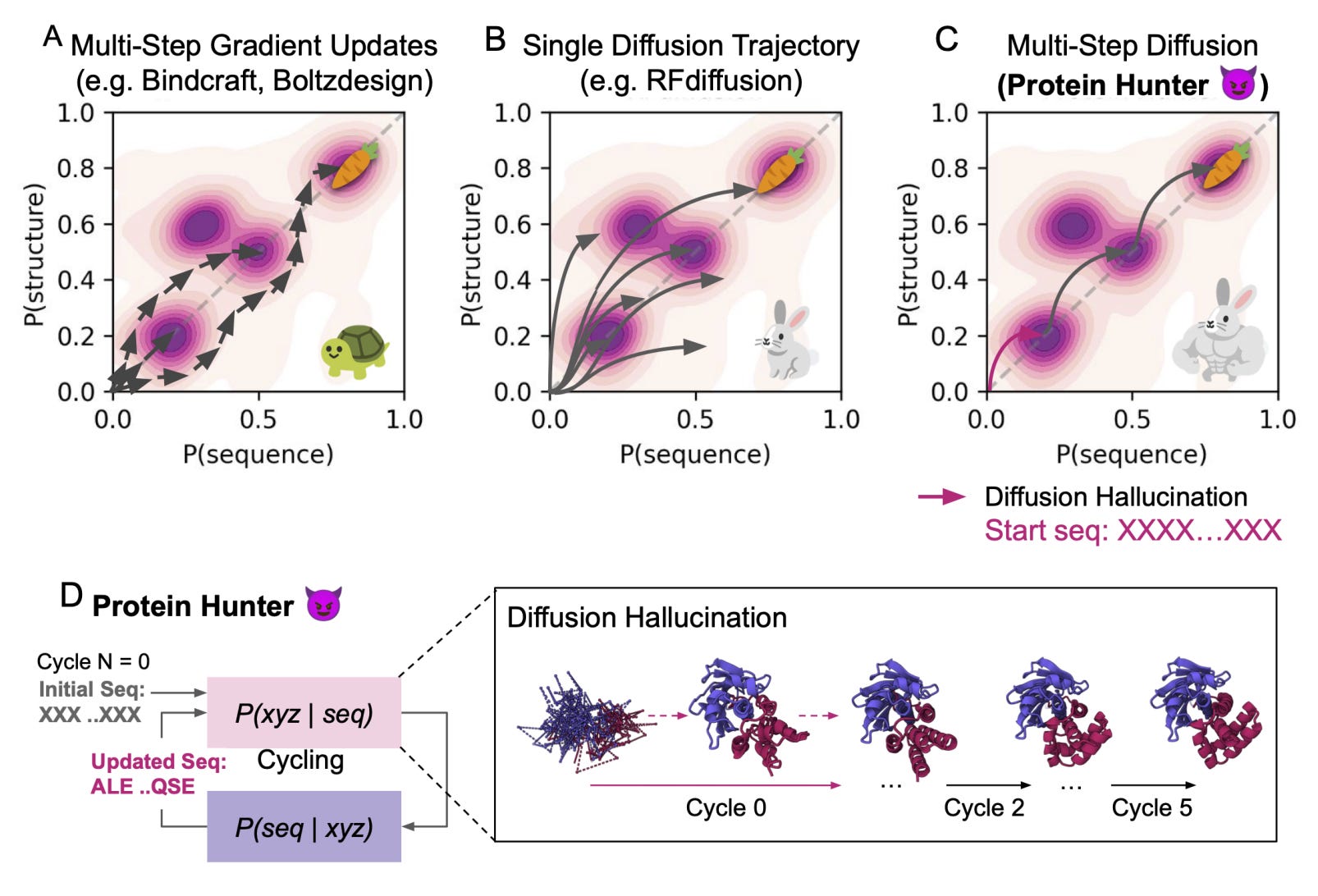
Applications and Insights
Zero-shot protein generation
From a blank sequence, Protein Hunter produces well-folded proteins between 100-900 residues in under two minutes, fine-tuning free, yet reaching AF3-style confidence levels.Generalised binder design
Designs binders for proteins, peptides, small molecules, DNA, and RNA, achieving higher in silico success rates across most targets than RFdiffusion or BoltzDesign.Multi-motif scaffolding and partial redesign
Can fix parts of existing structures (motifs, frameworks, or binding pockets) while regenerating the rest, enabling both de novo design and engineering of known proteins.Controlled fold diversity
By adjusting diffusion biases, researchers can steer designs toward β-sheet or α-helical topologies, breaking the helical bias seen in most generative models.
Protein Hunter shows that structure prediction models really can do more than predict; they can actually create. I think this is cool because it switches the narrative of diffusion “hallucination,” once seen as noise, into a design feature. Instead of optimising endlessly, it lets the model imagine structure and sequence together, refining through simple, fast cycles until they converge on something stable.
In today’s world, that’s a big step for generative biology, turning what was once a computational artefact into a creative tool for building real, functional proteins.
📄Check out the paper!
⚙️The code is due for release soon.
ChemRefine: Taking the pain out of running ML and quantum chemistry side by side.
Combining quantum chemistry and machine learning has always been powerful, but historically messy. Most workflows involve patching together tools, juggling formats, and tuning everything by hand.
ChemRefine, developed at the University of Texas at Dallas, claims to fix that. It’s an open-source platform that combines ML potentials and quantum chemistry into a unified, automated workflow.
At its core, ChemRefine links ORCA’s quantum chemistry engine with ASE’s MLIP library. So, you can run conformer searches, transition states, redox or excited-state workflows, and switch seamlessly between DFT and ML methods. It also includes ChemRefineGPT, a custom LLM that writes YAML configs and job scripts from plain-text prompts. You describe the workflow, and it builds it.
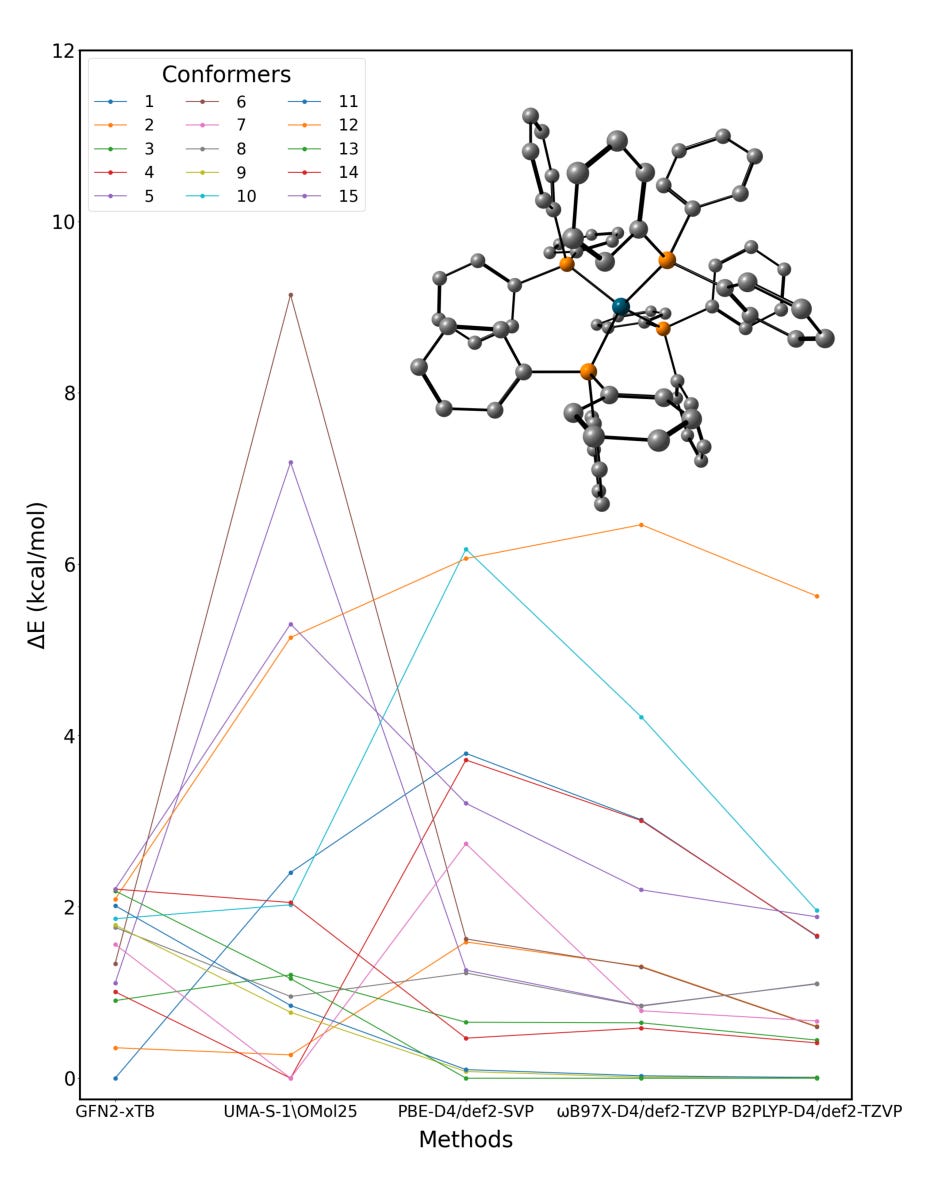
Applications and Insights
MLIP training and fine-tuning
Train models like MACE, UMA, or SevenNet using DFT data or normal-mode sampling. Get MD speeds 100x faster than DFT, with similar accuracy.Conformer refinement across theory levels
Automatically re-rank conformers using semi-empirical, hybrid, and double-hybrid methods to catch energy shifts missed at lower levels.Spin, redox, and excited-state analysis
Run TDDFT and spin-state workflows with seamless integration between MLIPs and electronic structure.TS and host-guest workflows
Find transition states, clean up vibrational modes, dock guests, and model microsolvation with ORCA’s built-in tools.
ChemRefine turns what used to be a mess of scripts into a single, intelligent platform for AI-assisted computational chemistry.
It brings the reasoning power of large language models together with the precision of quantum mechanics, making it easier to train, refine, and apply ML potentials across a wide range of chemical tasks.
It’s a look at what chemistry automation could become; not code-first, but idea-first.
📄Check out the paper!
⚙️Try out the code.
KERMT: Scaling Multitask Learning for Chemical Property Prediction
Fine-tuning large chemical models has become standard practice, but it’s still slow and fragmented. Most property prediction pipelines train one model per endpoint; accurate, but hard to scale when you’re tracking dozens of ADMET properties at once.
KERMT (Kinetic GROVER Multi-Task) changes that.
Developed by Meta AI and ETH Zurich, it’s a multitask extension of GROVER, a graph transformer pretrained on over 11 million molecules. The framework improves performance and efficiency across molecular property prediction tasks by introducing true multitask finetuning and new engineering optimisations for large-scale chemistry workloads.
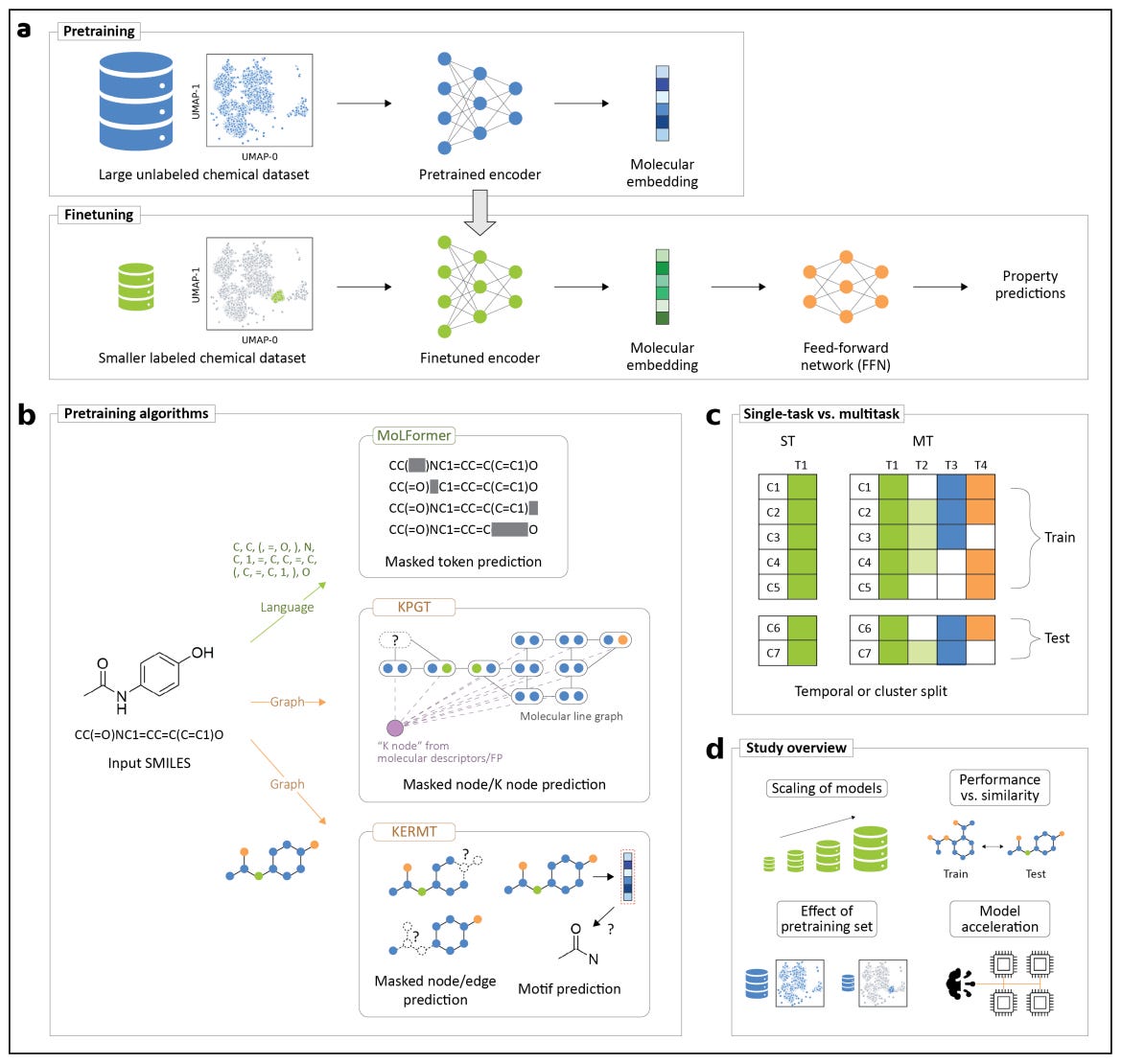
Applications and Insights
Multitask finetuning at scale
KERMT learns shared representations across related molecular properties, improving accuracy and data efficiency compared to single-task finetuning. The benefits are most pronounced in medium to large data regimes, where related tasks reinforce each other’s signal.Enhanced pretraining and architecture
Built on the GROVER backbone, KERMT uses atom- and bond-level message passing with motif and k-hop subgraph prediction tasks during pretraining, capturing both local and global structure information.Significant computational acceleration
The team reimplemented KERMT with distributed PyTorch (DDP) and integrated cuik-molmaker, a high-throughput molecular featurisation library. This setup achieved 2.2× faster finetuning and 2.9× faster inference, along with better GPU scaling efficiency and reduced CPU memory use.Robust multitask performance
On large ADMET datasets, KERMT outperforms non-pretrained GNN baselines and single-task models, showing clear gains in predictive correlation and stability. On public datasets like MoleculeNet, performance varies by task but KERMT remains competitive with other pretrained backbones.
I think this is cool because it shows how chemical foundation models are moving from individual tasks to shared infrastructure.
KERMT demonstrates that multitask finetuning isn’t just possible: it’s efficient, scalable, and production-ready. With distributed training, shared graph embeddings, and open datasets, it’s a practical step toward more unified, real-world molecular modelling pipelines.
Not another “bigger model.” Just a smarter way to use the ones we already have. Pretty nice.
📄Check out the paper!
⚙️Try out the code.
📊Public multi-task split dataset.
Thanks for reading!
Did you find this newsletter insightful? Share it with a colleague!
Subscribe Now to stay at the forefront of AI in Life Science.
Connect With Us
Have questions or suggestions? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website