
UW's RFdiffusion3, GRASP, and Stanford + Arc's GERMINAL
Kiin Bio's Weekly Insights

Welcome back to your weekly dose of AI news for Life Science!
We’ve had some feedback from readers asking for more detail on the data behind the models we feature.
Check out our new “What about the training data?” section for each article: covering what the model was trained on, where it came from, and how it was used.
What’s your biggest time sink along the drug discovery process?
RFDiffusion3: Protein Design at all-atom resolution
A new study from the University of Washington introduces RFdiffusion3 (RFD3), a generative model that builds proteins and their interactions in full atomic detail.
Earlier versions of RFdiffusion mainly worked at the residue level, with side chains added later. RFD3 changes that: it reasons over every atom across multiple chains and molecules, from proteins to DNA and small ligands. That shift makes it possible to design complete, functional interfaces in one step. Pretty exciting advancement.
The model is also reasonably leaner and faster, running about 10× quicker than RFdiffusion2 while keeping design quality high. Nice. With conditioning options like motifs, atom “hotspots,” or symmetry rules, RFD3 gives researchers fine control over tasks ranging from scaffolding epitopes to enzyme engineering.

Applications and Insights
1. All-atom, multi-chain generation
RFD3 generates both backbones and side chains simultaneously, capturing realistic contacts across proteins, DNA, and small ligands, giving a more complete view of molecular assemblies than earlier residue-level models.
2. Flexible conditioning
Design runs can be guided by motifs, symmetry, or atomic hotspots, letting researchers specify where binding should happen and how complexes should assemble. This makes it easier to design around functional constraints.
3. Greater diversity and foldability
Across benchmarks, RFD3 produced more unique and designable structures than earlier versions. Generated sequences folded back with AlphaFold more reliably, suggesting improved biological plausibility.
4. Experimental validation
DNA-binding proteins and enzymes designed with RFD3 were tested in the lab, with several showing measurable binding and catalytic activity: a strong sign that the improvements translate into real function.
What about the training data?
RFdiffusion3 doesn’t rely on massive pretraining outside structural biology. Instead, it is trained directly on curated protein complexes with full atomic detail:
Structure inputs come from the Protein Data Bank, covering protein–protein, protein-DNA, and protein-small molecule interactions. Additional AlphaFold2 distillation structures were used to expand coverage of folds and assemblies.
During training, native structures are noised at varying levels and the model is tasked with reconstructing backbone and side-chain coordinates under different conditioning signals like motifs, symmetry, or hotspots. This teaches the model to design realistic all-atom interfaces across diverse contexts.
All inputs are public, and the full code, weights, and evaluation pipeline are openly available in the RFdiffusion repository
I thought this was cool because it moves protein design from backbone sketches to full molecular assemblies. By generating everything at the atomic level and already producing experimental hits, RFD3 shows that generative biology is not just improving models: it is starting to deliver tools scientists can use at the bench.
📄Check out the paper!
⚙️The code will be available soon according to the authors!
GRASP: Integrating diverse experimental information to assist protein complex structure prediction
GRASP is a new tool for predicting protein complex structures which may be able to surpass AlphaFold3 in accuracy.
Setting itself apart from other structure prediction tools, GRASP combines experimental approaches, such as covalent labelling, crosslinking, chemical shift perturbation and deep mutational scanning to create accurate, 3D models of protein complexes. Exciting advancements.
These experimental datasets can be sparse and heterogenous, limiting their ability to be directly translated into structure prediction. However, Yuhao Xie alongside a team of researchers across various Chinese universities created GRASP: a framework allowing these experimental approaches to be used in conjunction with AlphaFold.

Applications and Insights
1. Integration of multiple data types
GRASP is able to take methods of experimental analysis and integrate them fluidly. For example, it can combine interface residues that have been identified from chemical shift perturbation with interaction surfaces that have been identified from covalent labelling, generating extremely accurate models of protein complexes.
2. Modelling interactions inside cells
GRASP can apply information about protein-protein interactions inside cells provided by crosslinking mass spectrometry to accurately model structures in situ.
3. Modelling antibody-antigen complexes
Hypervariable loops which form binding sites on antibodies make these complexes highly difficult to predict. AlphaFold also struggles to model these complexes because of conformational changes induced by binding. Incorporating information about interface residues from chemical shift perturbation allows GRASP to predict these structures more accurately.
4. Sparse experimental data + AI prediction
Experimental datasets can sometimes be incomplete, limiting their ability to model a complete 3D structure. GRASP uses AlphaFold to generate a baseline structure, then adds information from experimental datasets. This effective integration means that the model will have a higher degree of accuracy as opposed to one solely based on AI prediction.
GRASP shows how integrating AI prediction techniques with experimental data can create a powerful tool to model a protein complex with the highest degree of accuracy. Being able to model complicated structures such as an antibody-antigen complex demonstrates the value of integrating various approaches to structure prediction.
I thought this was cool because of its potential to aid the drug design process. Modelling complexes provides researchers with a basis to find binding sites, in order to develop inhibitors and other therapeutics with greater efficiency.
📄Check out the paper!
⚙️Try it out the code.
Germinal: Generate functional antibodies de novo with epitope-level control
A new study from Stanford University and the Arc Institute introduces Germinal, a generative framework that designs nanobodies and scFvs against specific epitopes with nanomolar affinities, while requiring only low-throughput experimental testing. This is exciting stuff.
Traditional antibody discovery relies on immunisation or huge screening campaigns. They work, but they’re expensive, slow, and offer no control over which part of the antigen is targeted. Germinal tackles this by co-optimising antibody structure and sequence: it combines AlphaFold-Multimer to enforce structural plausibility with an antibody-specific language model (IgLM) to ensure sequences look natural. This dual guidance steers designs toward realistic CDRs that hit the chosen epitope.
The team tested Germinal on four diverse antigens: PD-L1, IL3, IL20, and the viral protein BHRF1. Across just 43-101 designs per antigen, they achieved 4-22% experimental success rates, validating binders with nanomolar affinities that also expressed well in mammalian cells. That’s a major leap in efficiency compared to the thousands of designs usually required.
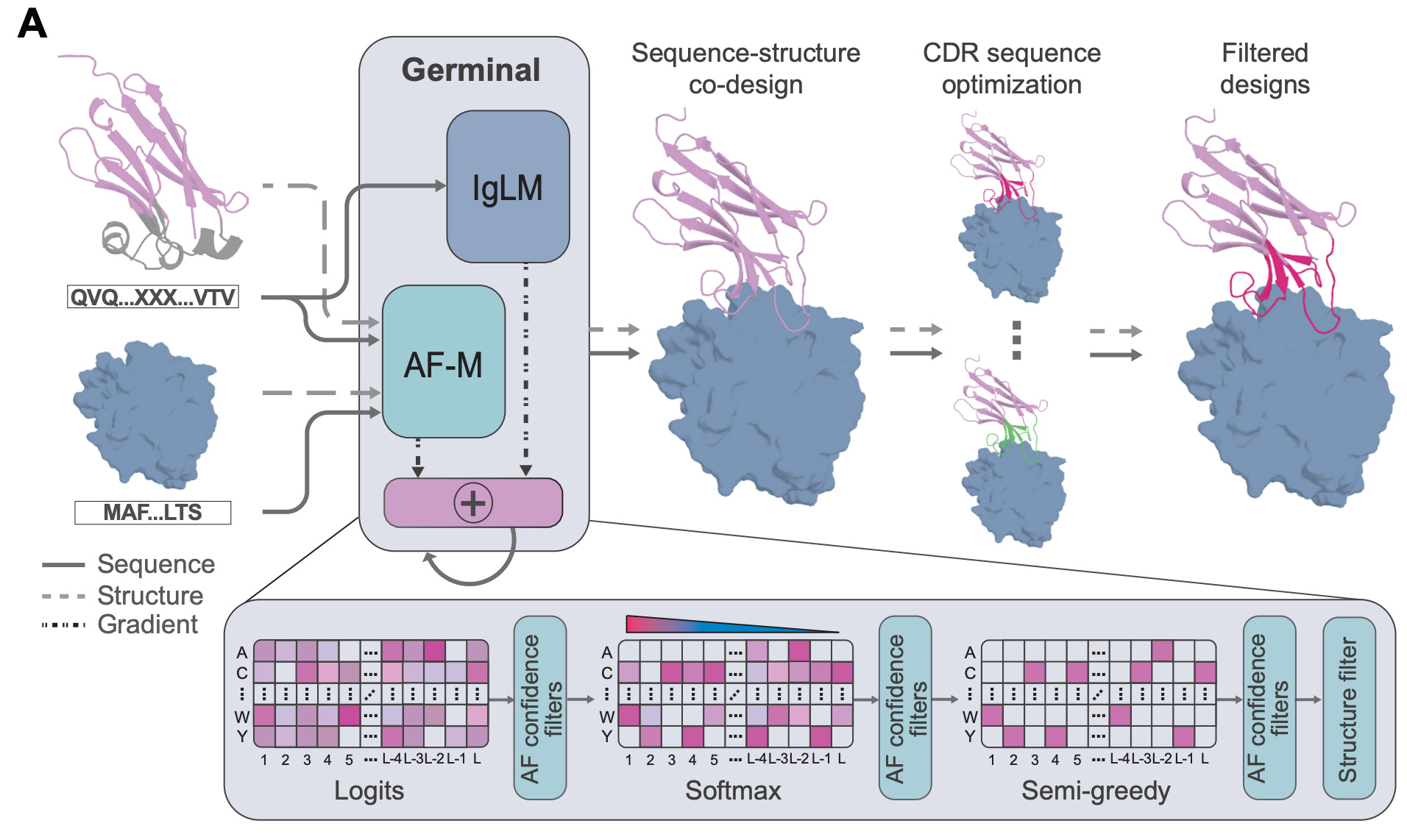
Applications and Insights
1. Efficient epitope targeting
Unlike library screening, Germinal can direct antibodies to user-defined epitopes, including functional or conserved regions that might otherwise be missed.
2. Low experimental burden
By narrowing libraries to a few dozen candidates, it cuts down validation cost and time. Binders emerged in days rather than months.
3. Maintains developability
Using natural frameworks and IgLM guidance, Germinal preserves expression and therapeutic-relevant properties while designing new CDRs.
4. Generalisable and open-source
The pipeline works for nanobodies and scFvs, scales across antigen types, and is fully open-source for the community.
What about the training data?
Germinal is trained with a combination of antibody-specific sequence data and structural priors:
Structure guidance comes from AlphaFold-Multimer, which ensures antibody–antigen complexes are structurally plausible and that candidate CDRs align with the target epitope.
Sequence guidance comes from IgLM, a language model trained on hundreds of millions of natural antibody sequences, providing realistic amino acid distributions and framework preferences.
During generation, Germinal proposes antibody structures against a chosen epitope, then iteratively filters them through IgLM scoring and AlphaFold-Multimer predictions. This balances novelty with developability.
All training data sources are public, and the full Germinal code and model weights are openly available for the community.
I thought this was cool because it solves one of the hardest problems in computational antibody design: generating new CDRs that actually bind without brute-force screening. Germinal shows that with smart integration of structure prediction and antibody language models, you can get real binders, fast, and with control over where they bind. That makes it a powerful tool for labs interested in molecular tools, therapeutics, or probing new epitopes. Very excited to see where Germinal takes the field.
📄Check out the paper!
⚙️Try it out the code.
Thanks for reading!
Did you find this newsletter insightful? Share it with a colleague!
Subscribe Now to stay at the forefront of AI in Life Science.
Connect With Us
Have questions or suggestions? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website