
Vilya Research's Vilya-1, TU Munich's TWIN, and Stanford's UCE
Kiin Bio's Weekly Insights

Welcome back to your weekly dose of AI news for Life Science! This weeks fix:
Vilya-1 predicts macrocycle conformations across diverse chemistries with a single all-atom model, beating physics-based methods and existing deep learning approaches on geometric accuracy. Macrocycles are notoriously hard to model, and this is the first foundation model built specifically for them.
TWIN is an implicit solvent ML potential trained on ab initio data that runs two orders of magnitude faster than explicit solvent while approaching DFT accuracy. If this holds up broadly, it removes one of the biggest bottlenecks in drug and peptide simulation.
UCE is now published in Nature after a 2023 preprint. A single-cell foundation model that embeds any cell type without retraining, trained on the Tabula Sapiens atlas. The zero-shot transfer results across tissues and species are what earned it the Nature publication.
Kiin Pioneer Programme
We built a platform that helps researchers speed up their entire science, from literature review and biomarker discovery to bioinformatics and computational chemistry. If your workflow involves pulling findings from five different places before you can actually act on any of them, this is for that.
The Pioneer Programme gives academic labs and non-profits one year of free access, plus support from our science team. No cost, no data transfer, all IP stays with your institution. Applications close August, cohort starts September.

Vilya-1: Macrocycle structure prediction foundation model
🧪 Where This Fits
Macrocyclic peptides occupy a sweet spot in drug design: large enough to hit protein-protein interactions that small molecules cannot reach, small enough to potentially cross cell membranes. The problem is that their conformational behaviour is extremely difficult to predict. They are too large for small-molecule force fields to handle well, too flexible for protein structure prediction methods, and too chemically diverse (cyclic, branched, non-natural amino acids, N-methylation) for any single modelling approach to cover.
Current options are either expensive physics-based sampling (molecular dynamics, metadynamics) that takes days per compound, or deep learning methods built for linear peptides that do not generalise to the cyclic, heavily modified structures that matter therapeutically. Tools like AlphaFold and OpenFold were not designed for this chemical space. Vilya-1 from Vilya Research is the first foundation model purpose-built for macrocycle structure prediction, using a unified all-atom representation that handles diverse topologies and chemical modifications in a single model.
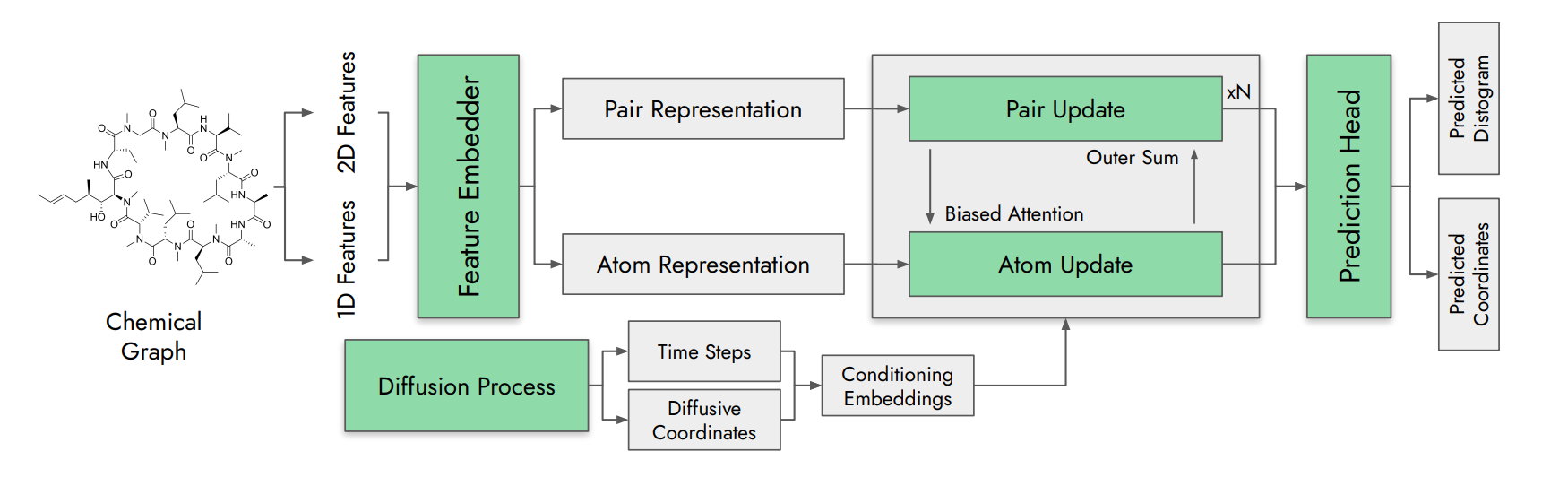
🔍 What It Is
Predicting macrocycle 3D structure is hard because they span a chemical space between small molecules and proteins that existing methods handle poorly. Sturmfels, Salem, Hiranuma et al. from Vilya Research present Vilya-1, a deep learning foundation model trained on heterogeneous structural datasets across diverse macrocycle topologies and chemistries.
Uses a uniform all-atom representation that accommodates cyclic peptides, non-natural amino acids, N-methylated residues, and other modifications without needing separate model architectures for each. Trained on a mix of experimental and computational structural data.
Improved geometric accuracy compared to physics-based methods and existing deep learning alternatives. Coverage extends to small molecules. Also supports predicting developability properties like membrane permeability, and generative design of novel macrocycles with specified properties.
💡 Why This Is Cool
Macrocycles are having a moment in pharma. Peptide drugs like semaglutide proved the market, and companies like Bicycle Therapeutics are building pipelines around constrained cyclic peptides. The structural prediction gap has been a real bottleneck: you can design a macrocycle computationally, but confirming it folds the way you expect requires either NMR (expensive, slow) or crystal structures (often impossible for flexible molecules). A model that gives you reliable conformational predictions for chemically diverse macrocycles would accelerate the design-make-test cycle considerably. The extension to permeability prediction is smart, since oral bioavailability is the other major hurdle for this drug class. No public code yet, which limits immediate evaluation by the community.
📃 Read the paper.
No public code repository available at time of writing.
TWIN: Transferable implicit solvent machine learning potential approaching ab initio accuracy
🧪 Where This Fits
Molecular dynamics simulations of drugs and proteins in solution face a fundamental trade-off: explicit solvent (modelling every water molecule) is accurate but extremely expensive computationally, while implicit solvent models (treating water as a continuum) are fast but sacrifice accuracy. For drug design, this means you either spend days simulating a single compound in explicit water, or you get fast results from implicit models that often disagree with experiment.
Previous implicit solvent approaches (GB/SA, COSMO) are based on empirical physics approximations that break down for many drug-like molecules. Recent ML potentials (ANI, MACE) have improved accuracy for gas-phase simulations, but extending them to solvation has been limited. TWIN from TU Munich takes an equivariant graph neural network, trains it exclusively on ab initio and experimental solvation data (no empirical force field data), and produces an implicit solvent potential that transfers across drugs, peptides, and proteins.
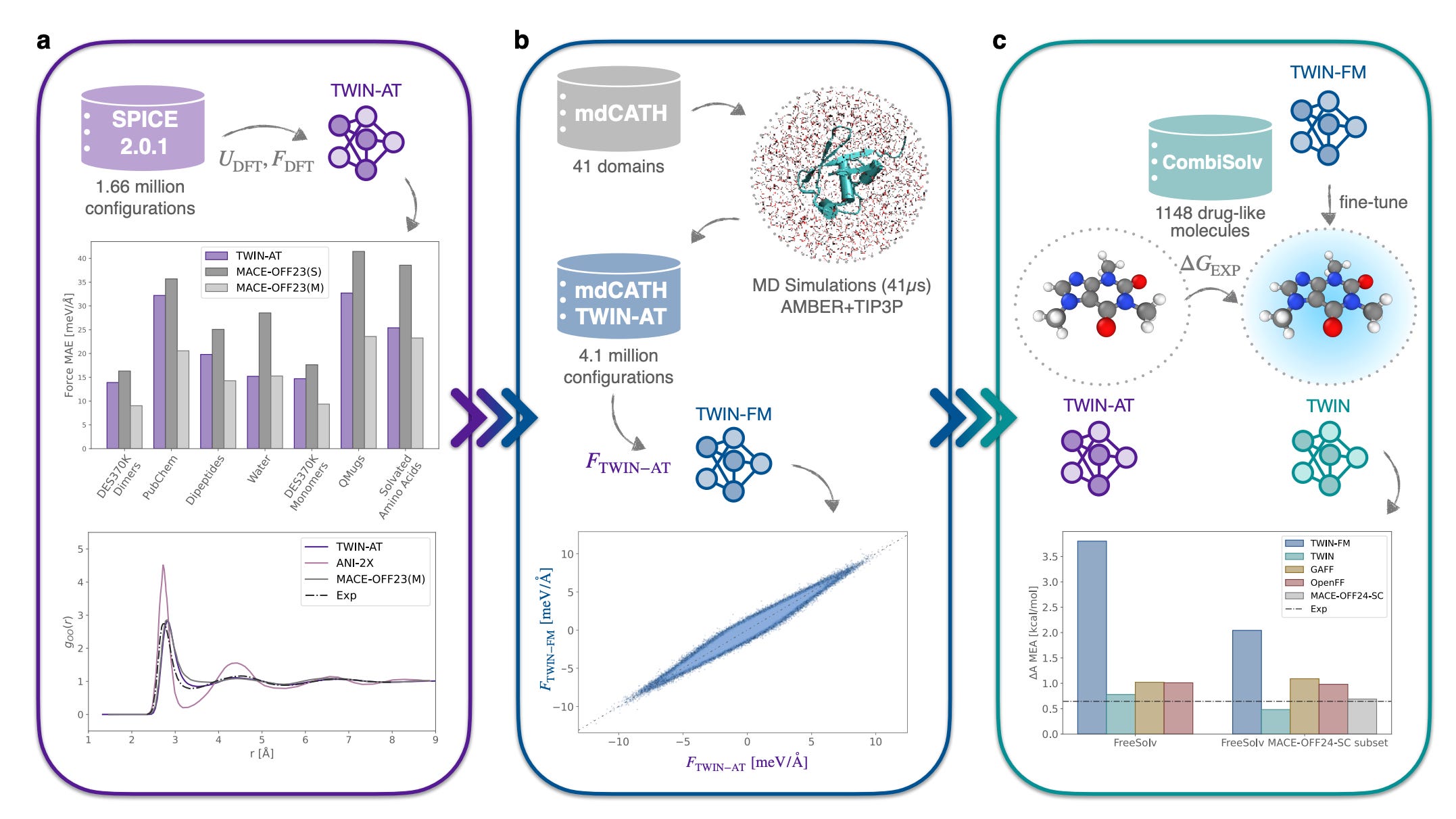
🔍 What It Is
Simulating molecules in water is either accurate and slow (explicit solvent) or fast and unreliable (implicit solvent). Eckwert and Zavadlav from TU Munich present TWIN (Transferable Water Implicit Network), an ML potential that combines the speed of implicit solvent with accuracy approaching DFT-level explicit solvent calculations.
Built on an equivariant graph neural network trained exclusively on ab initio quantum mechanical data and experimental measurements. Avoids any dependence on empirical force field parameters, which is what limits the transferability of traditional implicit solvent models.
Two orders of magnitude faster timestep evaluation than explicit solvent approaches. Outperforms prior ML-based implicit solvent models on crystallographic and NMR benchmarks. Transfers across drug molecules, peptides, and proteins without retraining.
💡 Why This Is Cool
Solvent is the hidden cost of computational drug design. Every binding free energy calculation, every conformational sampling run, every MD simulation spends most of its compute on water molecules rather than the drug you care about. If TWIN delivers on the promise of DFT-level solvation accuracy at implicit-solvent speed, and transfers reliably across chemical space, it could make free energy perturbation and metadynamics calculations accessible at scale rather than as expensive one-off studies. The key caveat: “approaching ab initio accuracy” on benchmarks does not always translate to “accurate enough for rank-ordering drug candidates.” The real test is whether medicinal chemists can trust it for prospective predictions. Two orders of magnitude speedup is the kind of change that alters what questions you can afford to ask.
📃 Read the paper.
No public code repository available at time of writing.
UCE: Universal cell embedding provides a foundation model for cell biology
🧪 Where This Fits
Single-cell foundation models have proliferated over the past two years: scGPT, Geneformer, scFoundation, and others all aim to learn general representations of cells from large-scale scRNA-seq data. The shared limitation is that most require fine-tuning on task-specific labelled data, and they struggle to generalise to cell types or tissues not seen during training. They also typically use gene-token vocabularies tied to a specific species, making cross-species transfer difficult.
UCE from Jure Leskovec and Stephen Quake’s groups at Stanford takes a different approach: it embeds cells using protein-level information (via ESM embeddings of gene products) rather than gene-name tokens, which makes the vocabulary species-agnostic. Trained on 36 million cells from the Tabula Sapiens atlas across multiple human tissues. The preprint appeared in late 2023, and the Nature publication confirms the approach held up through peer review with additional validation.
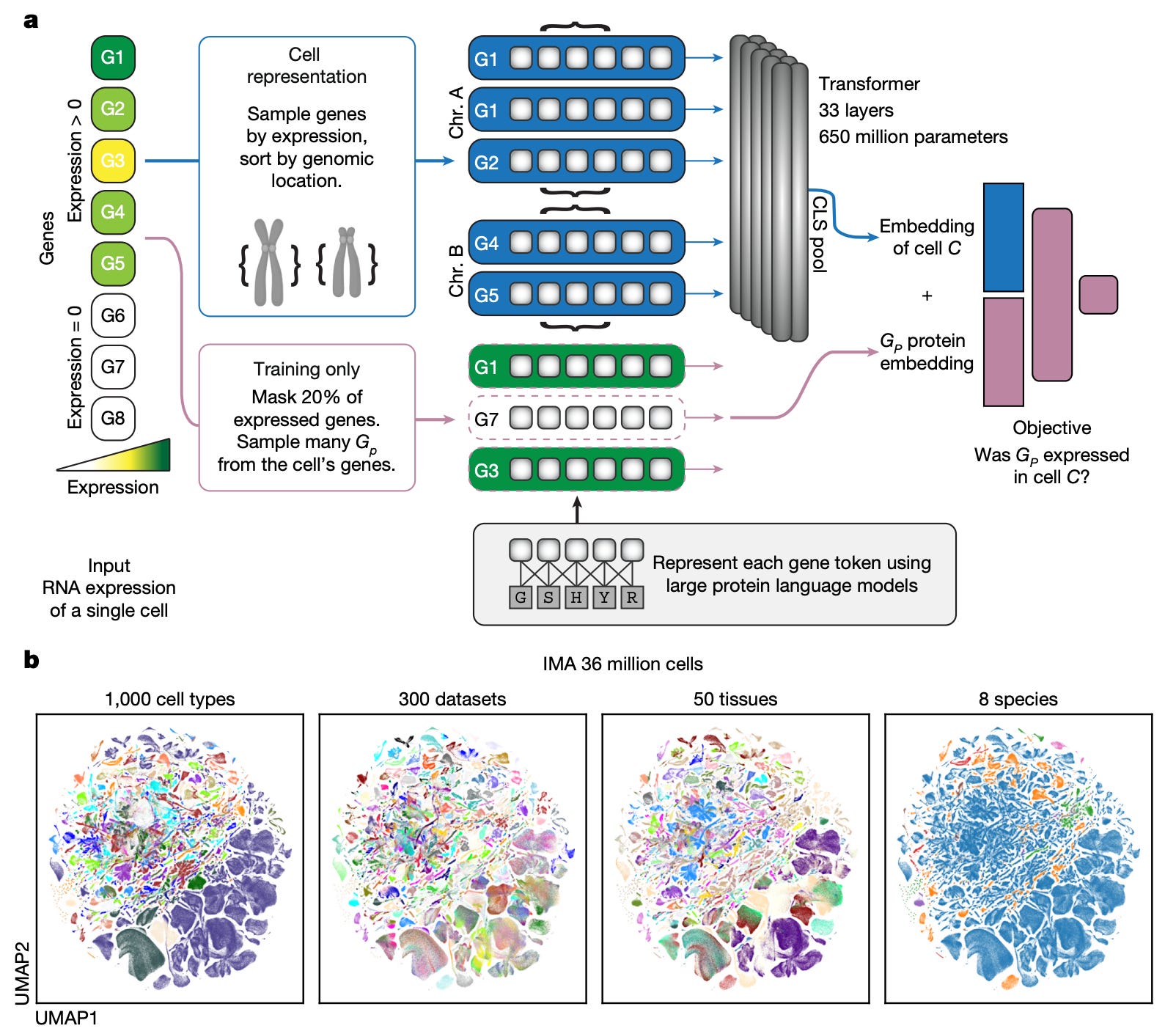
🔍 What It Is
Single-cell foundation models require fine-tuning for new tasks and do not transfer well across species or unseen cell types. Rosen, Roohani, Agrawal et al. from Stanford present UCE (Universal Cell Embedding), a foundation model that generates cell embeddings in a shared space without any task-specific retraining.
Uses ESM protein language model embeddings to represent genes (rather than arbitrary gene tokens), making the representation biologically grounded and species-transferable. Trained on 36 million cells across diverse human tissues from the Tabula Sapiens consortium.
Zero-shot cell type classification across tissues and species without fine-tuning. Embeddings capture biological relationships: similar cell types cluster together even across different organs and organisms. Published in Nature after extended peer review.
💡 Why This Is Cool
The protein-embedding approach is what makes this work where others have not. When you represent genes as ESM embeddings of their protein products, you get biological similarity for free: genes with similar functions have similar representations regardless of what organism they come from. That is why UCE can transfer across species without retraining, while models using gene-name tokens cannot. The Nature publication after a 2023 preprint suggests the reviewers were convinced this is not just a benchmark artefact. For single-cell researchers, the practical question is whether UCE embeddings are useful enough to replace the fine-tuning workflows they already have with scGPT or Geneformer. The 33-layer model and code are public, so that comparison is straightforward to make.
📃 Read the paper.
💻 Try the code.
🗓️ Events & Competitions
The best competitions, hackathons, and community challenges in AI x life sciences, curated weekly. Know something worth featuring? Reply and let us know.
More upcoming events:
BioHackathon Europe 2026 | November 9-13, Barcelona
ELIXIR’s annual international bioinformatics hackathon, running since 2018. 160+ participants, five days of collaborative coding on open bioinformatics infrastructure and tools. The call for project proposals has now closed.
Thanks for reading!
💬 Get involved
We’re always looking to grow our community. If you’d like to get involved, contribute ideas or share something you’re building, fill out this form or reach out to me directly.
Connect With Us
Have questions or suggestions? We'd love to hear from you!
📧 Email Us | 📲 Follow on LinkedIn | 🌐 Visit Our Website